Опубликовано в журнале:
«Атмосфера. Нервные болезни» 2007, № 2, с. 27-31Ишемический инсульт у больного, страдающего железодефицитной анемией
Б.А. Кистенев, М.Ю. Максимова, А.В. Лагутин
Клинический разбор Научного центра неврологии РАМННаиболее частыми факторами риска ишемических нарушений мозгового кровообращения (НМК) в среднем и пожилом возрасте, как известно, являются атеросклероз сосудов мозга и артериальная гипертензия [1]. Причины же ишемического инсульта у молодых пациентов значительно более многообразны и, как правило, связаны с относительно редкими заболеваниями и состояниями, что требует проведения целенаправленного диагностического поиска. Среди причин церебральной ишемии у больных молодого возраста важное место принадлежит разнообразным заболеваниям крови [2, 5].
В настоящей статье мы представляем случай ишемического инсульта, развившегося у пациента молодого возраста на фоне железодефицитной анемии.
Больной К., 37 лет, программист находился на обследовании и лечении в клинике Научного центра неврологии РАМН с 22.01.2007 по 28.02.2007 г
Из анамнеза известно, что пациент в течение 10 лет страдает геморроем. Три года назад начали беспокоить изжога, нарушение стула (чередование запоров и поносов), два года тому назад впервые почувствовал недомогание, общую слабость, быструю утомляемость, одышку при подъеме по лестнице на 3-й этаж. За медицинской помощью не обращался. В апреле 2006 г. больной амбулаторно сдал анализ крови, при котором впервые было выявлено наличие анемии, причем на протяжении 3–4 мес до этого ежедневно отмечал примесь алой крови в кале. С 24.04.2006 по 05.05.2006 г. обследовался в специализированном стационаре с диагнозом: “Железодефицитная анемия тяжелого течения на фоне хронического геморроя и частых геморроидальных кровотечений”. При поступлении отмечались жалобы на повышенную утомляемость, снижение работоспособности, одышку при нагрузках, плохое заживление ран и ссадин. При эзофагогастродуоденоскопии выявлены: рефлюкс-эзофагит с множественным эрозивно-пептическим поражением на фоне длинного сегмента желудочного эпителия в пищеводе (пищевод Баррета); гастро-эзофагеальная рефлюксная болезнь; атрофический гастрит. При колоноскопии диагностирован хронический геморрой. В анализах крови выявлено наличие анемии (гемоглобин 80 г/л). Проводилось лечение венофером в/в капельно, эритроцитарной массой, ферроплексом. В результате проведенного лечения состояние больного улучшилось: увеличилась физическая активность, перестала беспокоить одышка, уменьшилась общая слабость. Выписан под наблюдение терапевта и гастроэнтеролога. В период с мая по декабрь чувствовал себя удовлетворительно, принимал рекомендованный при выписке ферроплекс по 2 таблетки 3 раза в сутки в течение 3 мес.
Очередному ухудшению состояния предшествовали выраженные профессиональные перегрузки в течение 3 мес. 25 декабря 2006 г. на работе внезапно почувствовал резкую общую слабость, к которой затем присоединилась слабость в правой руке и ноге; добрался до дому на машине с посторонней помощью. Отмечено повышение артериального давления до 150/80 мм рт. ст., бригадой “скорой помощи” была сделана инъекция сульфата магния в/в струйно. До 27.12.2006 г. больной находился дома, причем деталей происходившего за этот период не помнит; со слов родственников, пациент с посторонней помощью с трудом добирался до туалета, наблюдалась выраженная неустойчивость при ходьбе.
С 27.12.2006 г. по 07.01.2007 г. находился на обследовании в неврологическом отделении городской больницы, где был установлен диагноз: “Острое нарушение мозгового кровообращения (ОНМК) в вертебрально-базилярной системе с формированием инфарктов в правой гемисфере мозжечка и правой половине моста. Железодефицитная анемия. Тромбоз интракраниального отдела позвоночной артерии справа. Артериальная гипертензия. Хронический геморрой”. В анализах крови вновь зафиксирован низкий уровень гемоглобина (73 г/л). При МРТ головного мозга (от 18.01.2007 г.) выявлена картина ишемических НМК в правой гемисфере мозжечка и правой половине моста головного мозга, а также смешанная гидроцефалия. Проводилось лечение антиоксидантами, ноотропными препаратами, препаратами железа, антиагрегантами. Для дальнейшего обследования был переведен в Научный центр неврологии РАМН.
При поступлении в клинику: общее состояние больного удовлетворительное, АД – 150/90 мм рт. ст.
В неврологическом статусе отмечается горизонтальный нистагм при взгляде в стороны, со стороны черепных нервов – без существенных нарушений. Определяется легкий правосторонний гемипарез с небольшим повышением тонуса по пирамидному типу, оживлением глубоких рефлексов и патологическими стопными знаками. В пробе Ромберга – выраженная мозжечковая атаксия. При ходьбе пошатывается, широко расставляет ноги в стороны. Координаторные пробы руками и ногами с двух сторон выполняет удовлетворительно. Правосторонняя гиперестезия. Тазовых расстройств нет. Высшие мозговые функции не нарушены.
Заключение нейроофтальмолога: гипертоническая ангиопатия сетчатки.
Заключение отоневролога: выявляется стволовая заднечерепная симптоматика.
ЭКГ: ритм синусовый, 76 ударов в 1 мин, нормальное положение электрической оси сердца.
Эхокардиография: пролапс митрального клапана 1-й степени с минимальной регургитацией.
Дуплексное сканирование магистральных артерий головы. Комплекс интима–медиа локально уплотнен и утолщен до 1,4 мм в бифуркации правой общей сонной артерии. Существенных структурных изменений в общих сонных, внутренних сонных и наружных сонных артериях не выявлено. Сонные артерии имеют прямолинейный ход. Показатели линейной скорости кровотока по сонным артериям в пределах возрастной нормы без значимой асимметрии сторон. В правой позвоночной артерии (диаметр 2,2 см) лоцируется низкий кровоток периферического типа – вероятнее всего, артерия заканчивается задней нижней мозжечковой артерией или окклюзирована в интракраниальном отделе. Левая позвоночная артерия имеет нормальный диаметр; кровоток по ней компенсаторно усилен. Асимметрия кровотока по позвоночным артериям около 70% (S > D).
Транскраниальная допплерография с детекцией микроэмболических сигналов: микроэмболических сигналов в течение 30 мин мониторирования кровотока по средним мозговым артериям не выявлено.
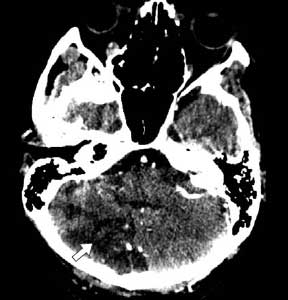
МРТ головного мозга: в правом полушарии мозжечка выявлен очаг измененного МР-сигнала (повышенной интенсивности в режиме Т2, пониженной – в Т1), неправильной формы, с четкими неровными контурами, без признаков объемного воздействия (рис. 1).
Рис. 1. Инфаркт в правом полушарии мозжечка (стрелка).
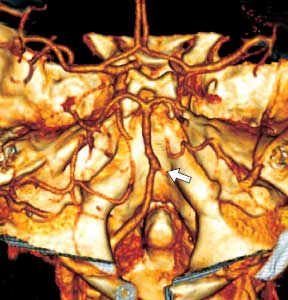
Компьютерно-томографическая ангиография: отсутствует кровоток по правой позвоночной артерии и по задней нижней мозжечковой артерии (рис. 2). Выявляется небольшой ретроградный заброс крови в дистальные отделы правой позвоночной артерии. Полученные КТ-данные соответствуют окклюзии правой позвоночной артерии.
Рис. 2. Окклюзия правой позвоночной артерии (стрелка).
МРТ шейного отдела позвоночника: патологии не выявлено.
КТ органов брюшной полости и забрюшинного пространства: органической патологии не выявлено.
Эзофагогастродуоденоскопия: эрозии тела желудка; пищевод Баррета.
Колоноскопия. Илеоцекальный клапан плоский, ориентирован в купол слепой кишки. Устье сомкнуто, округлой формы. В просвете кишки на отдельных участках выявляется умеренное количество содержимого черного цвета (пациент принимает препараты железа). Просвет ободочной кишки во всех отделах не изменен, тонус нормальный, циркулярные складки обычных размеров. Кишечная стенка эластична. Слизистая оболочка слепой, восходящей, поперечной ободочной, нисходящей и сигмовидной кишки розового цвета, с гладкой, блестящей поверхностью. Просвет прямой кишки не изменен, стенки эластичные, слизистая оболочка розового цвета. Сосудистый рисунок четкий.
Общий анализ крови (11.01.2007 г.): гемоглобин 95 г/л (норма 130–160 г/л), эритроциты 4,2 × 1012/л, цветной показатель 0,67; ретикулоциты 15–21‰; лейкоциты 5,5 × 109/л, сегментоядерные – 61%, палочкоядерные – 2%, эозинофилы – 3%, базофилы – 2%, лимфоциты – 24%, моноциты – 8%; тромбоциты – 266 × 109/л; СОЭ – 15 мм/ч.
Общий анализ мочи (11.01.2007 г.): удельный вес 1007, моча желтого цвета, мутноватая, среда кислая; эпителий плоский – единичный в редких полях зрения, лейкоциты – единичные в редких полях зрения; измененные единичные эритроциты изредка в препарате; слизь – немного.
Биохимический анализ крови (12.01.2007 г.): мочевина – 2,31 ммоль/л, общий белок – 70,3 г/л, глюкоза – 5,6 ммоль/л, холестерин – 3,9 ммоль/л, липопротеиды высокой плотности – 1,2 ммоль/л, липопротеиды низкой плотности – 1,9 ммоль/л, триглицериды – 3,12 ммоль/л (норма 0,45–1,7 ммоль/л), коэффициент атерогенности – 2,19; АЛТ – 28 Ед/л, АСТ – 18 Ед/л; КФК общая – 39 Ед/л, железо – 2,5 мкмоль/л (норма 10–30 мкмоль/л); билирубин общий – 6,49 мкмоль/л, билирубин прямой – отрицательный; креатинин – 96 ммоль/л.
Коагулограмма (12.01.2007 г.): время свертывания крови по Ли–Уайту – 9 мин 3 с, фибриноген – 3,9 г/л, гематокрит – 31%, фибринолитическая активность – 9%, индекс фибринолиза – 0,5; ретракция кровяного сгустка – 50%, протромбиновое время – 12,4 с, протромбиновый индекс – 79,7%, МНО – 1,035; тромбиновое время – 17,1 с, АЧТВ – 28,4 с, Б-фибриноген отрицательный, этаноловый тест отрицательный, Д-димеры <0,5 мкг/мл, антитромбин III – 60 и 139% в динамике при исследовании 05.02.2007 г. (норма 71–115%), протеин С – 124% (норма 69–115%), протеин S – 167% (норма 70–110%), антиген к фактору фон Виллебрандта – 150% (норма 61,3–117,5%); волчаночный антикоагулянт – 1,13 (норма 0,85–1,2), гомоцистеин – 10 мг/л (норма 8–10 мг/л), плазминоген – 101% (норма 73–126%).
Факторы свертывания крови (08.02.2007 г.): V фактор – 154%, VII фактор – 109%, VIII фактор – 220%, XII фактор – 132% (для всех исследуемых факторов норма 50–150%).
Агрегация тромбоцитов (11.01.2007 г.): под влиянием адреналина – 16%, под влиянием аденозиндифосфата (АДФ) – 31% (норма). Манжеточная проба: реакция адекватная (на фоне пробы происходило снижение агрегации тромбоцитов).
Исследование реологических свойств крови (12.02.2007 г.): вязкость крови и плазмы – в пределах нормы, индекс ригидности эритроцитов – 521 у.е. (норма 133 ± 16 у.е.), время образования двумерных агрегатов – 8,56 с (норма 7,5 ± 0,3 с), дезагрегация эритроцитов – в норме, средний объем эритроцитов – 68 фл (норма 80–100 фл), среднее содержание гемоглобина в эритроците – 21 пг (норма 30–35 пг). Таким образом, при данном исследовании выявлены микрогемореологические изменения (снижение деформируемости эритроцитов). Агрегация и дезагрегация эритроцитов в пределах физиологической нормы.
Анализ кала на скрытую кровь (08.02.2007 г. ) – отрицательный.
В отделении больному проводилось лечение тренталом, милдронатом в/в капельно, препаратами железа, хелатом железа, фолиевой кислотой, ранитидином. Выписан с улучшением состояния под наблюдение невролога и гематолога.
Таким образом, в данном случае у пациента молодого возраста развился тромбоз правой позвоночной артерии с формированием мозжечкового полушарного инфаркта. Это произошло на фоне железодефицитной анемии, повышения индекса ригидности эритроцитов и удлинения времени образования двумерных агрегатов, что указывало на имеющиеся гемореологические изменения. Можно предполагать, что появление ригидных форм эритроцитов связано с неэффективностью эритропоэза и образованием молодых и незрелых форм эритроцитов, характерных для анемии. При этом снижение деформируемости эритроцитов могло явиться фактором, инициирующим тромбообразование. Дополнительным фактором ОНМК такого генеза могли служить имевшие место повторные эпизоды повышения АД: как показывают многочисленные исследования, такие эпизоды обычно сопровождаются плазморрагиями и фибриноидным некрозом стенок артерий, что может осложняться набуханием стенок, резким сужением сосуда или закрытием просвета артерии [1].
Известно, что железодефицитные анемии – достаточно распространенные болезни, при которых снижается содержание железа в сыворотке крови, костном мозге и кровяных депо, в результате чего нарушается образование гемоглобина, а в дальнейшем и эритроцитов, возникают гипохромная анемия и трофические расстройства в тканях [2, 3]. Нередко железодефицитная анемия встречается у детей, подростков, женщин детородного возраста. Женщины страдают этим заболеванием значительно чаще, чем мужчины, хотя после 60 лет эта разница исчезает.
Наиболее частой причиной железодефицитной анемии являются кровопотери, особенно длительные и постоянные (при этом пусть даже и незначительные): организм теряет больше железа, чем получает из пищи. Значительную роль в развитии железо-дефицитных анемий занимают кровопотери из желудочно-кишечного тракта. Они являются самой частой причиной дефицита железа у мужчин и второй по частоте причиной у женщин. Такие кровопотери могут быть следствием геморроя, язвенной болезни желудка и двенадцатиперстной кишки, опухолей желудка и кишечника, дивертикулов различной локализации, глистных инвазий, эрозий слизистой оболочки желудка при грыже пищеводного отверстия диафрагмы [3]. В таких случаях препаратом выбора для лечения железодефицитной анемии становится Ферлатум – полусинтетический железо-протеиновый комплекс. Особенность Ферлатума заключается в преципитации белка вокруг атомов трехвалентного железа в кислой среде желудка, предотвращении контакта железа со слизистой и исключении раздражающего действия на ЖКТ. В слабощелочной среде кишечника происходит растворение белковой оболочки, высвобождение и всасывание железа.
Клиническая картина дефицита железа в организме очень многообразна и зависит от ряда факторов. При недостатке железа в организме анемия проявляется не сразу. Ей предшествует длительный период латентного дефицита железа с четкими признаками снижения запасов железа в организме без явных симптомов малокровия. При значительном снижении гемоглобина на первый план выступают симптомы, связанные с недостаточным обеспечением тканей кислородом: слабость, головокружение, сердцебиение, одышка, синкопальные состояния. Дефициту железа свойственны так называемые сидеропенические симптомы – изменения кожи, ногтей, волос (они не встречаются при других видах малокровия), а также мышечная слабость, не соответствующая глубине анемии, извращения вкуса (патофагия) и запаха (патоосмия). У больных часто наблюдаются сухость и трещины кожи на руках и ногах, ангулярный стоматит. Трещины в углах рта при дефиците железа бывают у 10–15% больных.
Наиболее характерный лабораторный признак железодефицитной анемии – микроцитарная гипохромная анемия, т.е. анемия с большим количеством незрелых форм эритроцитов. Содержание гемоглобина при железодефицитной анемии может колебаться от 20–30 до 110 г/л в зависимости от степени дефицита железа. Содержание эритроцитов может быть нормальным или сниженным до 1,5–2 × 1012/л. Пропорционально снижению числа эритроцитов снижается и уровень гематокрита. Обычно снижается средний объем эритроцитов до 60–70 фл при норме 85–90 фл [4]. Снижение количества эритроцитов можно объяснить как снижением пролиферативной активности ядерных эритроидных элементов, так и усилением неэффективного эритропоэза. Кроме того, имеются данные о некотором укорочении продолжительности жизни эритроцитов при железодефицитной анемии [3].
У представленного больного причиной железодефицитной анемии явились геморроидальные кровопотери. Дополнительно при эзофагогастро-дуоденоскопии были диагностированы рефлюкс-эзофагит с множественным эрозивно-пептическим поражением на фоне длинного сегмента желудочного эпителия в пищеводе (пищевод Баррета), гастроэзофагеальная рефлюксная болезнь и атрофический гастрит. В анализах крови отмечено стойкое снижение гемоглобина до 80 г/л.
Известно, что реологические свойства крови определяются многими факторами, однако наибольшее значение имеют деформируемость и агрегация эритроцитов. Изменения кислородотранспортной функции эритроцитов являются одним из важных механизмов приспособления к расстройствам кровообращения, а высвобождаемые ими факторы (АДФ и др.) способны оказать существенное воздействие на процессы внутрисосудистого тромбообразования [5]. При анемии вследствие уменьшения способности эритроцитов связывать, переносить и отдавать О2 развивается гемический тип гипоксии. При этом наиболее адекватным компенсаторным механизмом является увеличение скорости продукции эритроцитов в ответ на гипоксию. Именно этот механизм мобилизуется при постгеморрагической анемии и в большинстве случаев компенсирует потерю циркулирующих эритроцитов. Центральное место в механизме регуляции эритропоэза занимает почечный эритропоэтин, контролирующий и регулирующий эритроидную дифференциацию. При постгеморрагической анемии гипоксия тканей вызывает повышение образования эритропоэтина в почках в течение 4–6 ч, хотя из-за продолжительного периода от начала стимуляции эритроидных предшественников до освобождения ретикулоцитов из костного мозга компенсаторное увеличение числа циркулирующих эритроцитов начинается лишь через 4–5 сут. В крови обнаруживается абсолютное увеличение числа ретикулоцитов и появление нормобластов. Таким образом, увеличение уровня эритропоэтина при постгеморрагической анемии является важным физиологическим механизмом регуляции эритропоэза, направленным на улучшение снабжения тканей кислородом за счет увеличения в кровяном русле количества эритроцитов, включая молодые, незрелые клетки [2–4].
Недостаток железа, ограничивая уровень выработки эритроцитов количественно, ведет и к качественным изменениям в популяции эритроцитов. При железодефицитной анемии эрит-ропоэз в значительной мере бывает неэффективным за счет гибели большого количества эритроцитов в процессе их созревания и развития [7], при этом темп обновления популяции эритроцитов при хроническом течении железодефицитной анемии не возрастает, а падает. Одним из факторов, обусловливающих это, служит замедление синтеза ДНК в базофильных нормобластах у таких больных [3, 7].
Еще W. Duke в 1910 г. [8] обнаружил удлинение времени кровотечения у больных анемией и сделал заключение о влиянии эритроцитов на гемостаз. В 1960 г. A. Hellem [10] показал, что эритроциты содержат фактор R (позже описанный как АДФ), вызывающий агрегацию кровяных пластинок. Анализ данных литературы позволяет выделить два механизма активации тромбоцитов и гемостаза: 1) выделение из эритроцитов в плазму в результате их повреждения физиологического активатора тромбоцитов – АДФ; 2) физическое воздействие эритроцитов на тромбоциты, стимуляция их адгезии и агрегации [5, 6]. В механическом воздействии эритроцитов на тромбоциты также имеет значение деформируемость эритроцитов: ригидные эритроциты создают большой вращательный эффект в потоке крови и увеличивают вероятность столкновения тромбоцитов друг с другом и с сосудистой стенкой, приводя к травматизации и нарушениям функции эндотелия.
При повреждении эндотелия обнажается субэндотелиальный слой, кровяные пластинки контактируют с коллагеном, набухают и образуют псевдоподии. Тромбоциты уже в течение первых секунд активно прилипают к участку повреждения. Существенная роль в реакции тромбоцит–коллаген принадлежит специфическим мембранным рецепторам кровяных пластинок. Наряду со способностью фиксировать тромбоциты у места повреждения сосуда коллагеновые волокна инициируют высвобождение из пластинок экзогенных факторов агрегации. Одновременно с адгезией происходит образование агрегатов, содержащих от 3 до 20 тромбоцитов, под воздействием на них большого количества агрегирующих факторов – АДФ, адреналина, тромбина и др. [5]. Одновременно из тромбоцитов в кровь выделяются различные физиологически активные вещества, в том числе факторы свертывания, способствующие гемокоагуляции и вовлечению в процесс новых тромбоцитов, ретракции сгустка, что в целом обеспечивает надежное восстановление целостности сосудистой стенки [11]. Большинство гемостатических нарушений при железодефицитной анемии ассоциируется не только с нарушением целостности сосудистого эндотелия, но и с нарушением его функциональных возможностей [4].
Признание ведущей роли эритроцитов в процессах тромбогенеза при анемии не исключает, однако, значимости плазменных факторов. Одновременно с сосудисто-тромбоцитарным гемостазом включается и плазменный гемостаз за счет активации факторов свертывания крови, адсорбированных на поверхности тромбоцитов [11]. Так, у представленного больного отмечено повышение уровня фактора VIII до 220% (при норме 50–150%). Активация его при железодефицитных анемиях, по мнению ряда авторов, является компенсаторной реакцией в ответ на развитие повторных кровотечений [4, 11]. Вместе с тем повышение содержания фактора VIII характеризуется четким протромботическим эффектом и является доказанным фактором риска артериального тромбоза [12, 14–16].
Патологическое тромбинообразование предотвращается системой естественных антикоагулянтов, среди которых центральное место занимает антитромбин III. Он синтезируется в печени и эндотелиальных клетках, за счет чего эндотелий обладает высокой антикоагулянтной активностью и у здоровых людей абсолютно атромбогенен. На его долю приходится до 80% всей антикоагулянтной активности плазмы. Антитромбин III – плазменный α2-глобулин, блокирующий тромбин и другие активированные ферментные факторы свертывания – X, IX, XI, ХII, VII. Активируется антитромбин III связыванием с гепарин-сульфатом и глюкозаминогликанами, выстилающими поверхность эндотелия. Комплекс гепарин–антитромбин III фиксируется в значительном количестве на эндотелии, что играет важную роль в поддержании тромборезистентности интимы кровеносных сосудов [11, 13]. Имеются интересные данные о том, что антитромбин III является мощным вазодилататором мозговых артерий человека, обеспечивающим защиту от различных вазоконстрикторных эффектов [4]. У представленного пациента в остром периоде инсульта отмечалось снижение антитромбина III до 60%. По мнению большинства авторов, истощение антитромбина III при анемиях, обусловленное дисфункцией эндотелия, может провоцировать возникновение тромбозов и тромбоэмболий.
По данным литературы, НМК при железодефицитных анемиях встречаются в 1–2% случаев [9, 17]. Их особенностью является, с одной стороны, довольно быстрый регресс симптоматики (от нескольких часов до нескольких недель), с другой стороны – склонность к рецидивированию. При проведении КТ и МРТ у таких пациентов обычно выявляются очаги инфарктов, не соответствующие бассейну кровоснабжения какой-либо артерии. Наиболее часто очаги располагаются в затылочной, теменной и височной областях больших полушарий, реже – в лобной доле, мозжечке или базальных ганглиях; нередко они бывают множественными. Иногда повторные эпизоды НМК развиваются с интервалом в 1–3 мес в симметричных участках мозга.
Исходя из “эритроцитарного” патогенеза железодефицитных анемий оправданным в нашем наблюдении явилось применение трентала (пентоксифиллина), который является антиагрегантом с преимущественным влиянием на агрегационную активность эритроцитов [5, 18]. Немаловажно, что помимо хорошо известного гемореологического эффекта препарат угнетает вазоконстрикцию в микроциркуляторном русле, способствует уменьшению уровня фибрина плазмы и стимулирует фибринолиз. В низких дозах трентал действует также на циклооксигеназный путь, способствуя стимуляции синтеза и высвобождения простациклина и уменьшая продукцию тромбоксана [5]. Так реализуется антитромботическое и “эндотелий-сберегающее” действие препарата, в том числе при железодефицитной анемии.
Таким образом, можно заключить, что в представленном случае развившееся ОНМК ишемического характера в вертебрально-базилярной системе было обусловлено тромбозом правой позвоночной артерии в связи с изменением реологических свойств крови, гемостатической активацией и эндотелиальной дисфункцией сосудистой стенки на фоне железодефицитной анемии.
ЛИТЕРАТУРА
1. Верещагин Н.В. и др. Патология головного мозга при атеросклерозе и артериальной гипертонии. М., 1997.
2. Идельсон Л.Н. Гипохромные анемии. М., 1980.
3. Павлов А.Д. // Патологич. физиология и эксперим. терапия. 1991. № 4. С. 57.
4. Руководство по гематологии / Под ред. Воробьева А.И. М., 2005.
5. Суслина З.А. и др. Ишемический инсульт: кровь, сосудистая стенка, анти-тромботическая терапия. М., 2005.
6. Born G.V.R., Katzer M.A.A. // Acta Med. Scand. 1981. V. 210. Suppl. 651. P. 85.
7. Brunstrom G.M., Fielding J. // Br. J. Haemat. 1968. V. 14. P. 525.
8. Duke W.W. // JAMA. 1910. V. 60. P. 1185.
9. Hartfield D.S. et al. // Pediatr. Neurol. 1997. V. 16. P. 50.
10. Hillman R.S., Henderson P.A. // J. Clin. Invest. 1969. V. 48. P. 454.
11. Horne M.C. // Consultative Hemostasis and Thrombosis. Philadelphia, 2002. P. 15.
12. Koster T. et al. // Lancet. 1995. V. 345. P. 152.
13. Kraaijtnhagen R.A. et al. // Thromb. Haemost. 2000. V. 83. P. 5.
14. Meade T.W. et al. // Lancet. 1980. V. 1. № 8177. P. 1050.
15. O’Donnell J. et al. // Thromb. Haemost. 2000. V. 83. P. 10.
16. Rosendaal F.R. // Thromb. Haemost. 2000. V. 83. P. 1.
17. Yakushiji Y. et al. // Cerebrovasc. Dis. 2005. V. 20. P. 475.
18. Танашян М.М., Домашенко М.А. // Атмосфера. Нервные болезни. 2005. № 4. С. 21.
| Февраль 2010 г. |