Звукоизоляционные панели phonestar отзывы . . Плитка варан kerama marazzi купить в москве . |
Опубликовано в журнале:
«Неврология/ревматология, приложение consilium medicum» 2010, № 1, с. 22-29Взаимосвязь развития метаболических и когнитивных нарушений у пациентов с сахарным диабетом, предиабетом и метаболическим синдромом
В.Н.Шишкова
ГНИЦ Профилактической медицины, МоскваОдной из значимых причин развития когнитивных нарушений и деменции в настоящее время является сахарный диабет (СД). Когнитивные нарушения не всегда бывают следствием только структурного поражения головного мозга, их развитие может быть обусловлено метаболическими расстройствами, связанными с тяжелыми соматическими или эндокринными заболеваниями, или могут иметь место сочетания нескольких патологических факторов. В случае своевременной диагностики и лечения когнитивные нарушения, связанные с метаболическими расстройствами, могут полностью или частично регрессировать [1]. Важно знать патогенетические основы развития самых распространенных и социально значимых заболеваний современности, в этой иерархии СД занимает лидирующую позицию.
Распространенность
Распространенность СД во всем мире достигла эпидемических уровней и продолжает неуклонно возрастать. По данным Всемирной организации здравоохранения (ВОЗ), в 2000 г. в мире насчитывалось около 160 млн больных СД, эпидемиологи ВОЗ прогнозируют, что к 2025 г. численность больных СД превысит 400 млн человек, из которых 80-90% составят больные СД типа 2 (СД 2) [2]. Увеличение распространенности СД сопровождается повышением частоты сосудистых осложнений, которые являются причиной слепоты, хронической почечной недостаточности, ишемической болезни сердца (ИБС), инфаркта миокарда, инсульта, периферической полинейропатии и тяжелых нарушений функционирования центральной нервной системы (ЦНС), приводящих к ранней инвалидизации и высокой летальности таких пациентов.
Возраст
Возраст больных на момент дебюта заболевания СД 2 постепенно становится меньше, в частности в развивающихся странах, где максимум выявления диабета - от 45 до 64 лет (по сравнению с группой 65 лет и старше в развитых странах). Отмечается также тревожное повышение риска развития СД 2 у подростков и детей [3]. Раннее развитие СД, особенно у молодых людей, связано с более частой смертностью, разными осложнениями, приводящими к инвалидности, сниженной социальной активностью и низкому качеству жизни. Долгосрочные последствия этой эпидемии выливаются в огромные человеческие страдания и экономические затраты.
Осложнения
Исследования показали, что в дебюте СД 2 около 50% больных уже имеют макро- и микрососудистые осложнения. Возможно, это результат того, что метаболические нарушения возникают гораздо раньше первых клинических проявлений СД и к моменту постановки диагноза приводят к необратимым сосудистым изменениям. Наиболее драматично развитие центральных форм диабетической нейропатии, к которым относятся острые нервно-психические расстройства на фоне декомпенсации метаболизма (кетоацидоз, лактатацидоз гиперосмолярного и гипогликемического состояний), острые нарушения мозгового кровообращения - инсульт, преходящие нарушения мозгового кровообращения, прогрессирующая диабетическая энцефалопатия (ДЭ). К истинно диабетической принято относить прогредиентно развивающуюся на фоне нарушений углеводного обмена метаболическую энцефалопатию [1]. Однако выделение «чистой» метаболической формы энцефалопатии при СД весьма проблематично, поскольку с течением заболевания прогрессируют церебральные сосудистые нарушения, обусловленные развитием сосудистых изменений, артериальной гипертензии (АГ) и автономной нейропатии. ДЭ обычно развивается исподволь, у молодых пациентов ее хронические проявления усугубляются последствиями перенесенных острых гипер- и гипогликемических эпизодов, у пожилых - нарушениями мозгового кровообращения.
Когнитивный дефицит
Как и при других метаболических энцефалопатиях, клинические проявления ДЭ неспецифичны, наиболее часто развивается нарушение когнитивных функций: снижение памяти и внимания, замедление мышления, снижение скорости психических реакций и способности к обучению.
Установлена общность механизмов развития когнитивного дефицита при СД и болезни Альцгеймера, больные СД входят в группу риска развития деменции. Известно, что в процессе старения мозга принимают участие те же патогенетические механизмы, что и при развитии диабетических осложнений, ключевым из которых является оксидативный стресс. Выявляемые при нейропсихологическом обследовании изменения у больных СД 2 более стойкие, чем при СД типа 1 (СД 1). Это чаще всего средней степени выраженности нарушения вербальной памяти и процесса обработки информации. Иногда при СД выявляются нарушения праксиса, гнозиса, речевых и пространственных функций, зрительной и слуховой памяти, а также нарушения межполушарных взаимодействий с дисфункцией правого полушария [4].
Течение СД сопровождается частыми колебаниями уровня сахара в крови, порой резкими, которым придается очень большое значение в развитии мозговых расстройств. Особенно опасны в этом отношении гипогликемические эпизоды, возникающие во время терапии инсулином или пероральными сахароснижающими средствами.
Когнитивный дефицит часто имеет место и у больных с впервые диагностированным СД 2, еще не получавших сахароснижающую терапию, но у которых уже диагностируется энцефалопатия. Это состояние, по мнению многих ученых, при диабете является неизбежным [5]. Патогенез ДЭ представляет собой многофакторный процесс, сходный с таковым при диабетической полинейропатии, когда в патогенез оказываются вовлеченными сосудистая дисфункция, приводящая к уменьшению кровоснабжения нервов и мозговой ткани, нарушение трофики и прямое токсическое влияние гипергликемии на нервы. Влияние диабета на мозг более выражено у пожилых людей и приводит к ускорению обусловленного старением когнитивного снижения и развитию энцефалопатии [6].
Влияние инсулина
Особым вопросом в общей проблеме СД является влияние инсулина на когнитивные функции. Инсулин, идентичный панкреатическому, и типичные инсулиновые рецепторы широко экспрессируются в разных отделах мозга. Инсулин проникает в мозг посредством рецепторзависимого транспорта через гематоэнцефалический барьер и принимает участие в регуляции энергетического гомеостаза, репродуктивных и когнитивных функций организма. Вопросы, касающиеся нейрональных эффектов инсулина, остаются во многом дискуссионными, однако в последнее 10-летие получены убедительные доказательства того, что инсулин и инсулинрецепторная сигнальная система мозга необходимы для нормального функционирования нейронов. Дисфункция этой системы приводит к развитию нейродегенеративных заболеваний [7]. Было показано, что инсулин и инсулиновые рецепторы играют важную роль в синаптической передаче и могут быть связаны с такими важнейшими функциями мозга, как пищевое поведение, обучение и память. Гиперинсулинемия (ГИ) может определять когнитивное снижение, а нарушения в системе синтеза и секреции инсулина - негативно влиять на когнитивные функции [8]. Установлено также, что у больных СД 2, получающих инсулин, высок риск развития деменции, которая не просто отражает тяжесть диабета, но может быть и непосредственно связана с назначением инсулинотерапии [9].
Таким образом, с одной стороны, инсулинотерапия ослабляет токсический эффект хронической гипергликемии, с другой - у пациентов с СД 2 приводит к выраженной ГИ, что может обусловить прямое повреждающее действие инсулина на головной мозг в связи с нарастающими изменениями в синаптических структурах и клеточных мембранах и привести к развитию деменции. В настоящее время нет клинических исследований, где была бы доказана эффективность сахароснижающих препаратов, применяемых в современной терапии СД, особенно типа 2, в предотвращении развития именно когнитивных нарушений [10].
Изучение патофизиологических основ развития СД может дать представление о масштабах распространенности связанных с ним заболеваний и возможность их своевременной коррекции.
Развитие нарушений углеводного обмена и их связь с нарушениями жирового обмена
В крупных эпидемиологических исследованиях было убедительно показано, что диагностика патологических состояний гипергликемии только по уровню глюкозы крови натощак, как ранее рекомендовалось Американской диабетической ассоциацией, приводит к недооценке истинной распространенности диабета более чем на 1/3.
В настоящее время общепринятыми диагностическими лабораторными критериями СД являются пороговые уровни гликемии натощак и через 2 ч после нагрузки глюкозой (табл. 1).
Таблица 1. Диагностические критерии СД и других состояний гипергликемии
Патология Концентрация глюкозы
в венозной кровиСД ммоль/л мг/дл Натощак ≥7,0 126 Через 2 ч после приема глюкозы ≥11,1 200 НТГ Натощак <7,0 126 Через 2 ч после приема глюкозы 7,8-11,0 140-199 НГН Натощак 6,1-6,9 110-125 Через 2 ч после приема глюкозы <7,8 140 Согласно современным представлениям нарушенная толерантность к глюкозе (НТГ) и нарушенная гликемия натощак (НГН) являются состояниями, способными прогрессировать с исходом в СД 2. В 1990-х годах эксперты ВОЗ предложили использовать новый термин -«предиабет», который объединил НТГ и НГН. Сегодня в мире около 314 млн человек имеют предиабет, через 20 лет их число увеличится в 1,5 раза и составит около 500 млн человек Именно они пополнят многомиллионную армию больных СД 2 уже в ближайшем будущем.
Установлено, что частота развития СД у пациентов с НТГ и НГН примерно одинаковая. Поскольку НТГ более распространенное состояние в популяциях, чем НГН, то с этим состоянием связано большее число новых случаев СД. По данным эпидемиологических исследований, в группе пациентов с предиабетом переход в СД 2 зависит от возраста, расовой принадлежности, степени ожирения и в среднем составляет 596 в год (от 3,6 до 8,796). При 5-летнем наблюдении за такими пациентами СД 2 разовьется у 35-4096, а при сочетании НТГ и НГТ - у 6596 пациентов [6]. Поэтому изучению особенностей течения предиабета, его ранней диагностике и возможности первичной и вторичной профилактики придается большое значение. Врачу любой специальности рекомендуется выявлять факторы риска развития СД 2 (рис. 1) и проводить скрининг (табл. 2), поскольку рано выявленный предиабет и правильно предпринятые профилактические меры могут изменить судьбу пациента.
Рис. 1. Факторы риска развития СД 2.
*Применимо к лицам европеоидной расы.
Возраст 45 и более лет
Избыточная масса тела и ожирение (ИМТ≥25 кг/м2)
Семейный анамнез СД (родители и сибсы с СД 2)
Привычно низкая физическая активность
НГН или НТГ в анамнезе
Гестационный СД или рождение крупного плода в анамнезе
АГ ≥140/190 мм рт. ст.
Холестерин ЛПВП≤0,9 ммоль/л и/или триглицериды ≥2,82 ммоль/л
Синдром поликистозных яичников
Наличие ССЗТаблица 2. Скрининговые тесты: глюкоза плазмы натощак или ПГТТ с 75 г глюкозы
Возраст начала скрининга Группы, в которых проводится скрининг Частота обследования Любой возраст ИМТ>25 кг/м2 + 1 из факторов риска Лица с предиабетом - 1 раз в год >45 лет Нормальная масса тела в отсутствие факторов риска При нормальном результате - 1 раз в 3 года Главным фактором риска развития СД 2 у пациента является наличие избыточного веса, особенно при отложении жира в абдоминальной области. В последние 10-летия ученые стали рассматривать разные метаболические нарушения или заболевания, ассоциированные с избыточным весом, в комплексе и высказывать предположения об общности этих процессов. В 1960-е годы делались попытки объединения взаимосвязанных метаболических нарушений, ускоряющих развитие макрососудистых атеросклеротических заболеваний и СД 2. В 1988 г. американский ученый G.Reaven, объединив нарушения углеводного обмена, АГ и дислипидемию понятием «синдром X», впервые высказал предположение о том, что объединяющей основой этих нарушений может быть инсулинорезистентность (ИР) и компенсаторная ГИ [11]. В конце прошлого века метаболические нарушения и заболевания, развивающиеся у лиц с ожирением, объединили в понятие «метаболический синдром» (МС).
МС
МС - это сочетание метаболических нарушений, в патогенезе которых важную роль играет ИР, и которые являются факторами риска раннего развития атеросклероза и его сердечно-сосудистых осложнений (рис. 2). Согласно определению Международной федерации диабета, основными клиническими проявлениями МС являются:
- Абдоминальное ожирение.
- ИРиГИ.
- АГ
- Гипертриглицеридемия.
- Повышение холестерина (ХС) липопротеинов низкой плотности (ЛПНП) и снижение ХС липопротеинов высокой плотности (ЛПВП).
- НТГ и/или НГН натощак
- Нарушения системы гемостаза.
- Хроническое сосудистое воспаление.
Рис. 2. ИР как фактор риска развития сердечно-сосудистых заболеваний.

Существует 5 групп диагностических критериев МС. Отечественными учеными, экспертами Всероссийского научного общества кардиологов, также были разработаны и опубликованы критерии МС (2009 г.).
Основной критерий: центральный (абдоминальный) тип ожирения - окружность талии более 80 см у женщин и более 94 см у мужчин.
Дополнительные критерии:
- АГ (АД>130/85 мм рт. ст.);
- повышение уровня триглицеридов (>1,7ммоль/л);
- снижение уровня ХС ЛПВП (<1,0 ммоль/л у мужчин; <1,2 ммоль/л у женщин);
- повышение уровня ХС ЛПНП (>3,0 ммоль/л);
- гипергликемия натощак (глюкоза в плазме крови натощак 6,1 ммоль/л и более);
- НТГ (глюкоза в плазме крови через 2 ч после нагрузки глюкозой в пределах: >7,8 и <11,1 ммоль/л).
Наличие у пациента центрального ожирения и 2 дополнительных критериев является основанием для диагностирования у него МС.
Клиническая значимость нарушений и заболеваний, объединенных в рамки синдрома, заключается в том, что их сочетание в значительной степени ускоряет развитие и прогрессирование заболеваний, связанных с атеросклерозом, которые имеют не только медицинское, но и социальное значение в современном обществе. Кроме этого, многие исследователи рассматривают МС как прелюдию СД 2: риск развития СД 2 у лиц с МС в среднем в 5-9 раз выше, чем у лиц без него [12]. По данным статистики, у больных с впервые диагностированным СД 2 уже при 1-м обращении к врачу выявляются макро- и микрососудистые осложнения этого заболевания: нарушение зрения вследствие диабетической ретинопатии, нарушение функции почек вследствие диабетической нефропатии, поражение сосудов сердца, мозга, сосудов нижних конечностей и др. А при развившемся СД 2 риск развития сердечно-сосудистых заболеваний (ССЗ) в 3-4 раза выше, чем без него [13]. Именно эти осложнения являются основной причиной высокой ин-валидизации и смертности больных СД 2 - до 70% больных умирают от инфаркта миокарда или инсульта и их последствий.
Основные патогенетические механизмы развития ССЗ и СД 2 у пациентов с МС: роль ИР, ГИ и постпрандиальной гипергликемии
Ключевым звеном патогенеза МС является первичная ИР и компенсаторная ГИ.
ИР - это нарушение инсулинопосредованной утилизации глюкозы клетками. Это состояние, сопровождающее целый ряд физиологических и патологических процессов. Физиологическая ИР выявляется в пубертатном периоде, при беременности, в климактерическом периоде, во время ночного сна, при богатой жиром диете. Метаболическая ИР характерна для МС, СД 2, декомпенси-рованного СД 1, диабетического кетоацидоза, ожирения, выраженной недостаточности питания, гиперури-кемии, гипогликемии, индуцированной инсулином, злоупотреблением алкоголем. Эндокринная ИР отмечается при тиреотоксикозе, гипотиреозе, синдроме Кушинга, акромегалии, феохромоцитоме. Неэндокринная ИР типична для гипертонической болезни, хронической почечной недостаточности, цирроза печени, сердечной недостаточности, ревматоидного артрита, черного акантоза, миотонической дистрофии, травм, ожогов, сепсиса, состояния после хирургических вмешательств, раковой кахексии.
Наибольшее клиническое значение имеет потеря чувствительности к инсулину мышечной, жировой и печеночной тканями. Предполагают, что причиной ускоренного атерогенеза и высокой летальности от ИБС и инсультов у больных СД 2 также могут быть ИР и сопутствующая ей ГИ. У пациентов с ИР имеются дефекты генов, ответственных за передачу сигнала после соединения инсулина со своим рецептором (пострецепторные дефекты), и прежде всего у них нарушается транслокация и синтез внутриклеточного транспортера глюкозы ГЛЮТ-4. Но также могут быть генетические дефекты на уровне субстрата рецептора инсулина типа 1 и/или фосфатидилинозитол-3-киназы, также обнаружено нарушение экспрессии и других генов, обеспечивающих метаболизм глюкозы и липидов: глюкозо-6-фосфатдегидрогеназы, глюкокиназы, липопротеинлипазы, синтазы жирных кислот и др.
Генетическая предрасположенность к ИР может не реализоваться и не проявиться клинически (в виде МС и/или СД 2) при отсутствии необходимых для этого внешних факторов: избыточного калорийного питания (особенно жирной пищи) и низкой физической активности. Эти факторы сами по себе способствуют увеличению абдоминального ожирения, накоплению свободных жирных кислот (СЖК) и, следовательно, усилению имеющейся ИР. Развивающаяся при ИР компенсаторная ГИ, с одной стороны, позволяет в начале поддерживать углеводный обмен в норме, с другой - способствует развитию метаболических, гемодинамических и органных нарушений, приводящих в конечном итоге к развитию ССЗ и СД 2.
ИР мышечной ткани проявляется в снижении поступления глюкозы из крови в миоциты и ее утилизации в мышечных клетках. ИР жировой ткани проявляется в резистентности к антилиполитическому действию инсулина, приводящей к накоплению СЖК и глицерина. СЖК поступают в печень, где становятся основным источником формирования атерогенных липопротеинов очень низкой плотности. ИР ткани печени характеризуется снижением синтеза гликогена и активацией процессов распада гликогена до глюкозы (гликогенолиз) и синтеза глюкозы de novo из аминокислот, лактата, пирувата, глицерина (глюконеогенез), в результате чего глюкоза из печени поступает в кровоток В целом, ИР - это эволюционно закрепленный механизм выживания в неблагоприятных условиях, когда периоды изобилия чередовались с периодами голода. Наличие ИР обеспечивало накопление энергии в виде отложений жира, запасов которого хватало на то, чтобы пережить голод [14]. В современных условиях в странах с высоким экономическим развитием, постоянно сопутствующим изобилием и склонностью к малоподвижному образу жизни сохранившиеся в генетической памяти механизмы ИР продолжают «работать» на накопление энергии, что способствует развитию абдоминального ожирения, дислипидемии, раннего атеросклероза, АГ и СД 2.
Клинические исследования: трактовка результатов
К настоящему времени опубликованы результаты более 10 клинических исследований с участием не менее 15 тыс. человек, которые позволяют утверждать, что ИР и сопутствующая ей ГИ являются факторами риска ускоренного атерогенеза и высокой летальности от ИБС. Также достаточно клинических доказательств тому, что ГИ является независимым фактором риска развития ИБС у лиц без СД 2: Paris prospective Study (около 7 тыс. обследованных), Busselton (более 1 тыс. обследованных) и Helsinki Policemen Study (982 обследованных) [15]. В последние годы это подтверждено и у больных СД 2. Этим данным есть экспериментальное обоснование. Работы RStout свидетельствуют о том, что инсулин оказывает прямое атерогенное действие на стенки сосудов, вызывая пролиферацию и миграцию гладкомышечных клеток, синтез липидов в них и пролиферацию фибробластов. Таким образом, ИР и ГИ вносят весомый вклад в прогрессирование атеросклероза как у лиц без СД, так и у больных СД 2 [16].
Существенную роль играет ИР в развитии АГ. Взаимосвязь ГИ (маркера ИР) и эссенциальной АГ настолько прочна, что при высокой концентрации инсулина плазмы у больного можно прогнозировать развитие у него в скором времени АГ. Причем эта связь прослеживается как у больных с ожирением, так и у лиц с нормальной массой тела.
Существует несколько механизмов, объясняющих повышение артериального давления (АД) при ГИ (рис. 3). Инсулин способствует активации симпатической нервной системы (СНС), повышению реабсорбции Na и воды в почечных канальцах, внутриклеточному накоплению Na и Са. Инсулин как митогенный фактор активирует пролиферацию гладкомышечных клеток сосудов, что ведет к утолщению стенки сосудов. Механизм влияния инсулина на СНС до конца не ясен. Предполагают, что инсулин может активировать СНС путем прямого воздействия на ЦНС, проникая через гематоэнцефалический барьер в перивентрикулярную область гипоталамуса, где, связываясь со своими рецепторами на поверхности нейронов, блокирует активность парасимпатической нервной системы и, напротив, активирует СНС G.Reavean - основоположник синдрома ИР - предположил, что причиной гиперактивации СНС в условиях гипергликемии может быть повышенный метаболизм глюкозы в ядрах гипоталамуса, что тормозит передачу блокирующих импульсов на симпатические центры продолговатого мозга.
Рис. 3. Роль ИР в патогенезе АГ.
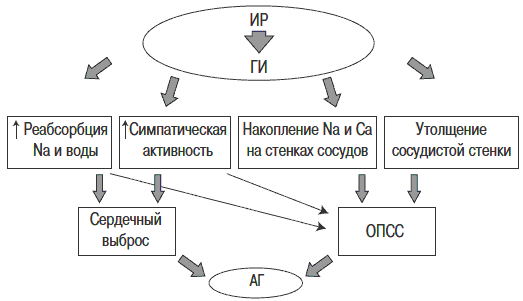
Стимуляция СНС при ГИ сопровождается увеличением сердечного выброса, повышением общего периферического сосудистого сопротивления (ОПСС), что неизбежно приводит к повышению АД. Одновременное снижение активности парасимпатической системы, вызванное ГИ, увеличивает частоту сердечных сокращений. Повышение реабсорбции Na и воды происходит также под влиянием ГИ. Инсулин оказывает прямое воздействие на проксимальные канальцы нефронов, повышая реабсорбцию Na и жидкости. Помимо антинатрийуреза инсулин вызывает антикалийурез и антиурикозурию. В результате увеличивается объем циркулирующей жидкости, что приводит к повышению сердечного выброса.
Внутриклеточное накопление Na и Са - эффект действия инсулина. Инсулин блокирует активность Na-K- и Ca-Mg-АТФазы клеточных мембран, что приводит к повышению внутриклеточного содержания Na и Са. Вследствие накопления этих электролитов в стенке сосудов повышается чувствительность сосудистых рецепторов к действию сосудосуживающих факторов. Под влиянием инсулина происходит утолщение стенки сосудов. Митогенные свойства инсулина обнаружены достаточно давно в серии экспериментальных работ, где было показано, что инсулин стимулирует клеточный рост, пролиферацию и миграцию гладкомышечных клеток сосудов, приводит к утолщению их стенки. В норме инсулин, связываясь с рецепторами на поверхности клеток эндотелия, может действовать двумя разными путями (рис. 4).
Рис. 4. Атерогенные и антиатерогенные свойства инсулина.
Примечание. shc – адаптивный белок; ras – белок, связывающий shc; raf – киназа серин-треонина; МАРК – митоген активированной протеинкиназы; IRS-1 и IRS-2 – субстраты инсулиновых рецепторов 1 и 2; PI3-K – фосфатидилинозитол-3-киназу, NO – оксид азота.
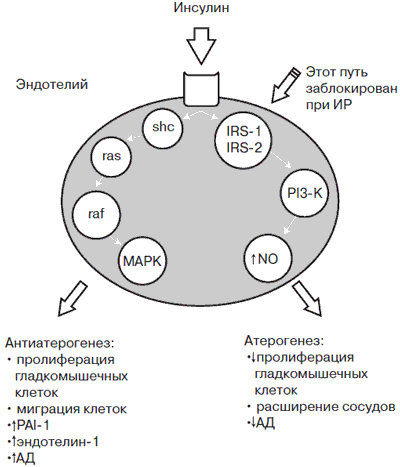
Первый путь - это активация секреции оксида азота (NO) через субстраты инсулиновых рецепторов 1 и 2 (IRS-1 и IRS-2) и фосфатидилинозитол-3-киназу (PI3-K). Этот механизм обеспечивает сосудорасширяющие и антиатерогенные свойства инсулина, участвует в инсулинзависимом транспорте глюкозы в клетки. Второй путь -реализация митогенных свойств инсулина через каскад посредников (адаптивный белок - she, белок, связывающий she - ras, киназа серин-треонина - raf), повышающих активность митогенактивированной протеинкиназы (МАРК), что завершается пролиферацией и миграцией гладкомышечных клеток, активацией синтеза сосудосуживающего фактора эндотелина-1 и повышением АД. Оказалось, что в условиях ИР 1-й механизм не работает - именно этот путь резистентен к действию инсулина, следовательно, молекула NO не синтезируется. В то же время 2-й механизм сохраняет свою высокую активность. Поэтому ГИ, развивающаяся вследствие ИР (при МС, СД 2, висцеральном ожирении), не только не снижает АД, а напротив, оказывает гипертензивное и атерогенное действие.
Существует взаимосвязь между активностью ренин-ангиотензиновой системы (РАС), уровнем АД и чувствительностью тканей к инсулину. Хорошо известно, что гиперактивность РАС стойко поддерживает высокое АД. Однако лишь недавно, в экспериментальных условиях, получены убедительные данные о том, что ангиотензин II (АТП) дозозависимо ингибирует пострецепторную сигнальную систему инсулина (комплекс IRS-1 и IRS-2, связанный с PI3-K), реализующую транспорт глюкозы в клетки и продукцию NO [16]. Одновременно АТП стимулирует МАРК, задействованную в осуществлении митогенной и пролиферативной активности инсулина.
Таким образом, гиперактивность РАС и АТП вызывает резистентность тканей к антиатерогенному и гипотензивному действию инсулина, что приводит к развитию ССЗ, а также блокирует транспорт глюкозы в клетки, что способствует развитию предиабета, а затем и СД 2.
Возможность профилактики развития СД 2
Проспективное исследование UKPDS (UK prospective Diabetes Study) позволило проанализировать прогрессирование СД 2 в течение более 10 лет и установило, что к моменту клинического дебюта заболевания только 50-60% от всей массы р-клеток поджелудочной железы продолжает активно секретировать инсулин - это 1-я причина, ведущая к заболеванию (рис. 5) [17]. Таким образом, метаболические нарушения, ведущие к развитию СД 2, реально развиваются задолго до клинического дебюта диабета. Примерно за 5-6 лет до манифестации диабета (при снижении функциональной способности р-клеток до 75%) можно диагностировать предиабет -НТГ или сочетание НТГ и НГН.
Рис. 5. Эволюция функции β-клетки поджелудочной железы.

Второй основной причиной развития СД 2 является сниженная чувствительность мышечной и жировой ткани, а также печени к действию эндогенного инсулина. Когда у пациента развивается клиническая картина СД 2 - это значит, что ИР тканей, которая существовала у него задолго до дебюта заболевания, уже привела к тому, что эндогенных запасов инсулина перестало хватать на преодоление существующей ИР. Функциональная активность р-клеток поджелудочной железы уже снизилась к этому времени на 50%, что и привело к повышению уровня гликемии.
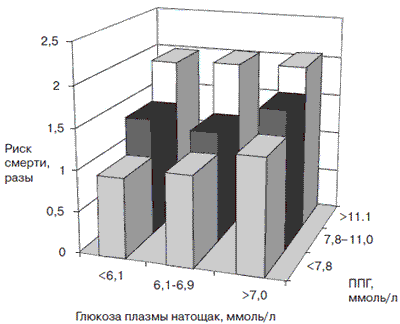
Долгое время (иногда в течение нескольких лет) субъективно больной может не ощущать признаков гипергликемии, т.е. заболевание протекает бессимптомно. В то же время сосудистые осложнения СД, в том числе и энцефалопатия, уже начинают развиваться, что обусловлено быстрым прогрессированием постпрандиальной гипергликемии. Уровню глюкозы плазмы крови после приема пищи (постпрандиальная гликемия) придается большое значение в прогрессировании сердечно-сосудистой заболеваемости. Постпрандиальная гипергликемия является самостоятельным фактором риска развития сердечно-сосудистых осложнений и преждевременной смерти, как показали результаты крупных международных исследований [18]. В исследовании DECODE использовались данные ряда проспективных крупномасштабных исследований, проводившихся в Европе. На основании их анализа было показано, что более высокий риск сердечно-сосудистой смертности имели пациенты с повышением постпрандиального уровня глюкозы более чем 11,1 ммоль/л, независимо от уровня гликемии натощак (рис. 6).
Рис. 6. Риск смерти от ССЗ в зависимости от уровня глюкозы. Примечание. ППГ - постпрандиальная гликемия.
Предупреждение осложнений СД
Начало СД 2 характеризуется не только выраженными нарушениями чувствительности тканей к инсулину (ИР), но и недостаточностью функции р-клеток поджелудочной железы (недостаточной для того, чтобы преодолеть ИР путем повышения секреции инсулина). Эти изменения на стадии клинических проявлений СД, безусловно, необратимы, поэтому даже раннее начало лечения СД 2 не может привести к полному выздоровлению, но применение лекарственных средств, влияющих на ослабление основных патогенетических влияний - в основном ИР, необходимо на любом этапе развития углеводных нарушений. В то же время, если начать профилактические мероприятия до дебюта СД 2 - на этапе предиабета, то можно предотвратить развитие и прогрессирование СД 2 и сопутствующих ему тяжелых осложнений. Идеи профилактики развития СД, как и начальные этапы современной терапии, опираются на одни и те же подходы и препараты, поскольку обусловлены патогенезом заболевания.
Рекомендации немедикаментозного плана включают изменение образа жизни, сочетающее соблюдение диеты и регулярные физические нагрузки, которые должны быть направлены на снижение массы тела, ИР, нормализацию АД, липидных нарушений и восстановление углеводного обмена.
В случае низкой приверженности пациентов изменению образа жизни, а также неэффективности немедикаментозных методов коррекции, оправдано применение препаратов, улучшающих чувствительность тканей к инсулину, т.е. облегчающих поступление глюкозы в голодающую клетку без дополнительной ГИ и, следовательно, прерывающих патогенетические связи развития и СД, и его осложнений.
Важно помнить, что при СД 2 самым эффективным методом является сочетание медикаментозной профилактики с изменением образа жизни.
Профилактика развития деменции у пациентов с нарушениями углеводного и жирового обменов
Профилактика любого осложнения, связанного с СД, особенно типа 2, - задача не из простых. Она связана с многими факторами: сложный патогенез заболевания и длительное латентное течение СД, не проявляющегося клинически первые 7-8 лет; требующиеся от врача, чаще всего невролога или терапевта, глубокие знания проблемы для проведения дополнительных методов обследования и ранней диагностики заболевания или направления к эндокринологу.
Таким образом, в современной практике чаще всего пациенту устанавливается диагноз СД 2 одновременно с появлением у него 1 -го макроваскулярного осложнения - острого инфаркта или инсульта, которое и является поводом для более серьезного дообследования.
Одной из первых при СД происходит поражение ЦНС и ПНС в виде периферической полинейропатии и энцефалопатии смешанного генеза - сосудистого и метаболического, что приводит к прогрессирующему снижению когнитивной функции и развитию деменции. А если учесть, что начавшаяся терапия СД тоже может оказывать неблагоприятный эффект на когнитивные функции пациента, то профилактика развития поражений головного мозга должна быть более активной.
Поэтому предупреждение развития деменции у таких пациентов прежде всего предполагает раннюю диагностику первых когнитивных нарушений - на стадии умеренных когнитивных расстройств (УКР). УКР - приобретенные объективные нарушения одной или нескольких когнитивных функций в результате органического поражения головного мозга, не приводящие к утрате пациентом независимости и самостоятельности в повседневной жизни. При УКР отсутствует бытовая, социальная и профессиональная дезадаптация, однако могут отмечаться затруднения при осуществлении сложных видов деятельности и обучении [19]. Для своевременной коррекции УКР препаратом выбора может считаться агонист дофаминовых рецепторов с дополнительным норадренергическим действием пирибедил (Проноран), который можно применять в качестве монотерапии или комбинации с другими средствами (нейротрофические, нейрометаболические средства, предшественники ацетилхолина).
Фармакологические особенности и клиническая эффективность
Механизм действия Пронорана связан с активацией D2/D3 дофаминовых рецепторов в лимбической системе и лобной коре, а также с блокадой а2-адренорецепторов, способствующей усилению норадренергической передачи в лимбической системе и лобной коре. Кроме того, как показывают экспериментальные данные, Проноран, блокируя а2-адренорецепторы, приводит к усилению высвобождения ацетилхолина в лобной коре и дорсальном гиппокампе, что также может вносить вклад в усиление когнитивных функций.
При УКР лечение Пронораном сразу же начинают с эффективной дозы 50 мг/сут. У большинства больных клинический эффект препарата становится очевидным к концу 1 -го месяца лечения, однако у части пациентов положительная реакция проявляется в более поздние сроки - на 2-м или 3-м месяце. Соответственно, рекомендуемая продолжительность лечения должна быть не менее 3 мес. Однако если к началу 2-го месяца приема препарата эффект отсутствует, дозу препарата рекомендуется повысить до 100 мг/сут. Простая схема применения и хорошая переносимость препарата в используемых дозах является залогом высокой комплаентности пациентов при лечении Пронораном.
В ряде исследований показано, что Проноран в дозе 50-100 мг/сут у больных с УКР способствует уменьшению выраженности когнитивных нарушений, связанных с лобной дисфункцией, параллельно стабилизируя аффективный статус и уменьшая такие субъективные проявления, как головная боль, головокружение, нарушение сна, утомляемость. Так, в 2004-2005 гг. в России было проведено широкое эпидемиологическое исследование ПРОМЕТЕЙ, направленное на выявление частоты когнитивных нарушений среди амбулаторных неврологических пациентов [20]. В рамках этого всероссийского исследования изучался ноотропный эффект Пронорана при его использовании в широкой амбулаторной практике как в виде монотерапии, так и в комбинации с вазоактивными и метаболическими препаратами. В исследовании участвовали 3210 пациентов, из них 1670 женщин и 1540 мужчин в возрасте от 60 до 96 лет, средний возраст составил 68,8±6,1 года. Показаниями к назначению Пронорана служили легкие или умеренные когнитивные нарушения возрастного и/или сосудистого характера, диагностика которых базировалась на жалобах пациента и данных объективного клинико-психологического исследования с применением MMSE и теста рисования часов. Проноран назначался в дозе 50 мг/сут в течение 12 нед, как в моно-, так и в комбинированной терапии (в сочетании с сосудистыми и метаболическими препаратами).
На фоне приема данного препарата было отмечено достоверное улучшение когнитивных функций по данным нейропсихологических методов исследования, по объективному клиническому впечатлению лечащего врача и собственному мнению пациента. Улучшение фиксировалось уже на 6-й неделе приема Пронорана и продолжало нарастать по выраженности к 12-й неделе лечения. По оценке врачей улучшение когнитивных функций имело место в 82% случаев, различий в эффективности моно- и комбинированной терапии отмечено не было. Помимо благоприятного ноотропного эффекта терапия Пронораном, по данным исследования ПРОМЕТЕЙ, оказывала положительное влияние в отношении субъективных симптомов дисциркуляторной энцефалопатии. На фоне терапии пациенты отмечали регресс выраженности таких симптомов, как головная боль, несистемное головокружение, шум в голове, нарушения сна, утомляемость и пониженное настроение. Уменьшение выраженности указанных симптомов отмечалось как на фоне монотерапии Пронораном, так и на фоне комбинированной терапии. Однако следует подчеркнуть, что комбинированная терапия не имела преимуществ перед монотерапией Пронораном.
Включенные в исследование пациенты были старше 60 лет, большинство из них имели одно или несколько сопутствующих заболеваний. Полученные результаты убедительно свидетельствуют о хорошей переносимости препарата пожилыми людьми. Нежелательные явления отмечались у небольшой части пациентов, не угрожали их жизни, не наносили непоправимого вреда здоровью, крайне редко требовали отмены проводимого лечения.
Таким образом, Проноран оказывает положительный ноотропный эффект при ранних недементных УКН сосудистой этиологии, которые свойственны пациентам с СД. Эффективность в отношении когнитивных расстройств, а также удовлетворительные профили переносимости и безопасности у пациентов пожилого возраста позволяют рекомендовать Проноран для широкого клинического применения при дисциркуляторной энцефалопатии у пациентов с СД.
ЛИТЕРАТУРА
1. Калинин АЛ., Котов СВ. Неврологические расстройства при эндокринных заболеваниях. М.: Медицина, 2001.
2. Wild S, Roglic G et al. Global Prevalence of Diabetes. Estimates for the year 2000 andprojections for2030. Diabetes Care 2004; 2 7:104 7-53-
3. Дедов ИИ, Ремизов О.В., Петеркова В А. Сахарный диабет второго типау детей и подростков. Сахарный диабет. 2001; 4:26-31.
4. Stewart R., Liolitsa D. Type 2 diabetes mellitus, cognitive impairment and dementia. Diabetic Med 1999; 16 (2): 93-112.
5. Vanhanen M., Koivisto K, Karjalainen L. et al. Risk for non-insulin-dependent diabetes in the normoglycaemic elderly is associated with impaired cognitive function. Neuroreport 1997; 8 (6): 1527-30.
6. Дедов ИИ., Шестакова МБ. Сахарный диабет. М: Медицинское Информационное Агентство, 2005.
7. Бондарева В.М., Чистякова О.Б. Инсулин и инсулинре-цепторная сигнальная система мозга. Нейрохимия.Ака-демиздатцентр «Наука»РАН. 2007; 24: 8-20.
8. Kalmijn S, Feskens EJ, Launer LJ et al. Glucose intolerance, hyperinsulinaemia and cognitive function in a general population of elderly men.Diabetologia 1995; 38 (9): 1096-102.
9. OttA, Stolk RP, van Harskamp F et al. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999; 53 (9): 1937-42.
lO. Areosa SA, Grimley EV. Effect of treatment of type II diabetes mellitus on the development of cognitive impairment and demencia. Cochrain Database SystRev 2002; 4-' CD003804.
11. Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988; 37: 1595-607.
12. Чазова И.Е., Мычка BE. Метаболический синдром. М.: Media Medica, 2004.
13. D'Agostino R, Hamman R, Karter A et al. Cardiovascular disease risk factors predict the development of type 2 diabetes. Diabetes Care 2004; 27 (9): 2234–40.
14. Амеrican Diabetes Association. Insulin resistance. Diabetes Care 1998; 21: 310–14.
15. Balkau B, Eschwege E. Insulin resistance: an independent risk factor for cardiovascular disease. Diabetes, Obesity Metab 1999; 1 (Suppl. 1): S23–31.
16. Nigro J, Osman N, Dart A et al. Insulin resistance and atherosclerosis. Endocrine Reviews 2006; 27 (3): 242–59.
17. U.K. Prospective Diabetes Study Group. U.K. Prospective Diabetes Study 16: Overview of 6 years'therapy of type II diabetes: a progressive disease. Diabetes 1995; 44: 1249–58.
18. Stern M, Fatehi P, Williams K et al. Predicting future cardiovascular disease. Diabetes Care 2002; 25 (10): 1851–6.
19. Яхно Н.Н. Когнитивные расстройства в неврологической клинике. Неврол. журн. 2006; 11 (1): 4–12.
20. Захаров В.В. Всероссийская программа исследований эпидемиологии и терапии когнитивных нарушений в пожилом возрасте («Прометей»). Неврол. журн. 2006; 11: 27–32.
| Октябрь 2012 г. |