Опубликовно в:
Вестник педиатрической фармакологии и нутрициологии № 2 2006Значение метаболических нарушений в генезе кардиомиопатий и возможности применения L-карнитина для терапевтической коррекции
И.В Леонтьева, В.С.Сухоруков
Московский НИИ педиатрии и детской хирургии Росздрава
Обследованы 75 детей с кардиомиопатиями, в возрасте от 2-х до 14 лет. Из них 30 больных с дилатационной кардиомиопатией, 26 больных с гипертрофической кардиомиопатией, и 19 больных с митохондриальными синдромами: Кернса-Сейра (n=7), Барта (n=1), MELAS (n=2), MERRF (n=1), гистиоцитарной кардиомиопатией (n=1), карнитиновой кардиомиопатией (n=3), с органическими ацидемиями, сопровождаемыми кардиомиопатией (n=4). Установлено, что для генетических синдромов при первичной митохондриальной патологии, характерен клинический полиморфизм поражения сократительного миокарда по типу дилатационной или симметричной гипертрофической кардиомиопатии в сочетании с нарушениями в проводящей системе сердца и развитием жизнеугрожаемых тахи- или брадиаритмий.Результаты работы показывают, что метаболические нарушения, связанные с митохондриальной дисфункцией, являются важным патогенетическим аспектом развития кардиомиопатий. Развитие метаболических изменений при кардиомиопатиях могут быть обусловлены как первичным, так и вторичным нарушением митохондриальных функций.
Установлено, что митохондриальная дисфункция при кардиомиопатиях поддается терапевтической коррекции. Применение L-карнитина для лечения кардиомиопатий позволяет улучшить функциональное состояние миокарда, способствует устранению сердечной декомпенсации, снижает показатели смертности.
Ключевые слова: дети, кардиомиопатии, лечение, L-карнитин, Элькар
Кардиомиопатии у детей относятся к тяжелой форме патологии миокарда. Они характеризуются злокачественным течением, резистентностью к проводимой терапии, высокой смертностью [1-4]. В настоящее время ведется активный поиск значения нарушений клеточной биоэнергетики в этиопатогенезе заболеваний миокарда [5-8].
Кардиомиопатии могут наблюдаться на фоне типичных митохондриальных синдромов, таких как синдром MELAS (митохондриальная миопатия - энцефалопатия - лактат-ацидоз - инсультоподобные эпизоды) [9], синдром MERRF (миоклонус-эпилепсия и наличие «рваных красных волокон» (ragged red fibers, RRF) в скелетных мышцах) [10], синдром Кернса-Сейра [11,12], митохондриальная миопатия [13]. Вместе с тем известно, что митохондриальная дисфункция может быть причиной развития не только полиорганной патологии, но и проявляться преимущественным поражением миокарда. Так, описаны случаи гипертрофической кардиомиопатии при дефиците цитохром-С-оксидазы [5,8], дефиците карнитина [14-15], снижении активности I, IV или II и III комплексов цепи дыхательных ферментов митохондрий [16], низкой активности пальмитоил-коэнзим-А-дегидрогеназы, связанной со снижением функции ацетил-коэнзим-А-дегидрогеназы длинных цепей [17].
В связи с этим, было выдвинуто предположение, что нарушения клеточной энергетики вследствие митохондриальной дисфункции могут являться важным аспектом патогенеза кардиомиопатий. Был предложен новый термин митохондриальные кардиомиопатии [5,8]. К ним были отнесены заболевания миокарда, сопровождающиеся структурными, количественными и функциональными нарушениями митохондрий, либо комбинацией этих нарушений, в основе которых лежат мутации митохондриальной ДНК.
В отечественной детской кардиологической практике митохондриальные кардиомиопатии при жизни диагностируются недостаточно достоверно или остаются нераспознанными. Отсутствуют критерии дифференцированного подхода к обследованию больных с кардиомиопатиями для выявления митохондриальной недостаточности, не разработаны пути метаболической коррекции нарушений клеточной энергетики.
Цель исследования:
выявить клинико-патогенетическую значимость нарушения клеточной энергетики при кардиомиопатиях у детей; предложить пути фармакологической коррекции нарушений энергетического обмена.
Пациенты и методы
В отделе кардиологии Московского НИИ педиатрии и детской хирургии Росздрава были обследованы 75 детей с кардиомиопатиями (40 девочек и 35 мальчиков), в возрасте от 2-х до 14 лет (средний возраст 7±2,1 лет). Из них 30 больных с дилатационной кардиомиопатией, 26 больных с гипертрофической кардиомиопатией, и 19 больных с митохондриальными синдромами: Кернса-Сейра (n=7), Барта (n=1), MELAS (n=2), MERRF (n=1), гистиоцитарной кардиомиопатией (n=1), карнитиновой кардиомиопатией (n=3), с органическими ацидемиями, сопровождаемыми кардиомиопатией (n=4).
Программа исследования включала электрокардиографию, рентгенографию грудной клетки, суточное холтеровское мониторирование ЭКГ, допплер-эхокардиографию, электромиографию, электроэнцефалографию (по показаниям). Уровень общего карнитина определяли энзиматическим методом с использованием наборов «Roche», Germany. Уровень лактата определялся по методу Баркера и Саммерсона, пирувата – методом Мюллера в модификации Л.Ф. Марченко на фоне стандартного глюкозотолерантного теста.
Морфологический анализ биоптатов скелетной мышцы проводился с применением световой гистохимии для выявления гликогена, кальция, липидов, определялась активность митохондриальных ферментов (сукцинатдегидрогеназы, цитохромоксидазы), также использовалась электронная микроскопия скелетной мышцы.
Результаты и обсуждение
Были проанализированы клинические варианты и степень выраженности изменений со стороны сердечно-сосудистой системы при митохондриальных заболеваниях.
При синдроме Кернса-Сейра выявлен широкий клинический полиморфизм нарушений со стороны сердечно-сосудистой системы. Во всех случаях изменения затрагивали проводящую систему сердца. Выраженность изменений в проводящей системе сердца широко варьировала от дистального нарушения проведения по правой или левой ножки пуска Гиса до полной атриовентрикулярной блокады. Поражения проводящей системы сердца носили непрерывно прогрессирующий характер, начинаясь с дистальной блокады ножек пучка Гиса, распространяясь на атриовентрикулярный узел с развитием полной атриовентрикулярной блокады. Возникновение полной атриовентрикулярной блокады приводило к резкой брадикардии, длительным паузам сердечного ритма с развитием синкопальных состояний — приступов Морганьи-Адамса-Стокса. Эти изменения требовали немедленной имплантации электрокардиостимулятора для профилактики жизнеугрожаемых состояний. Нарушения проводящей системы сердца при синдроме Кернса-Сейра в 5 из 8 случаев сочетались изменениями в миокарде по типу как дилатационной (3 из 8 наблюдений), так и гипертрофической симметричной необструктивной кардиомиопатии (2 из 8 наблюдений). Наши данные подтвердили мнение других авторов [18] о том, что развитие выраженной систолической левожелудочковой дисфункции в виде резкого снижения контрактильной способности миокарда (диффузный гипокинез стенок левого желудочка, уменьшение фракции укорочения левого желудочка и скорости циркулярного сокращения волокон), наличие обширных ишемических изменений резко ухудшают прогноз течения заболевания.
Под нашим наблюдением находились 2 пациента с синдромом MELAS (Митохондриальная миопатия - энцефалопатия - лактат-ацидоз, инсультоподобные эпизоды). В основе патогенеза синдрома лежат точковые мутации митохондриальной ДНК. В одном наблюдении наследственность отягощена. Старший брат пробанда умер в возрасте 10 лет (отмечались атрофия зрительного нерва, периодическая рвота, частые мигренеподобные головные боли, выраженная мышечная слабость, утомляемость, косоглазие), у матери частые головные боли, непереносимость физических нагрузок. Возраст дебюта заболевания в том и другом случае относился к 8 годам. Первые симптомы заболевания носили экстракардиальный характер: частые мигренеподобные головные боли, сопровождающиеся тошной и рвотой. Изменения неврологического статуса характеризовались мышечной слабостью, повышенной утомляемостью при физической нагрузке, координационными нарушениями (пошатыванием при ходьбе, неуверенностью при выполнении координационных проб). Впоследствии развились атаксия, нарушения речи (дизартрия, моторная дислалия), слуха (двусторонняя нейросенсорная тугоухость) и зрения (частичная атрофия зрительного нерва, расходящееся косоглазие, птоз левого века). В дальнейшем появились эпизоды тошноты, рвоты, снижения аппетита, усилились мигренеподобные головные боли, возникли эпилептические приступы с потерей сознания. Частота приступов в течение года нарастала, и в 12 лет возник инсульт, с развитием стойкого левостороннего гемипареза. На электроэнцефалограмме определялась пароксизмальная активность в виде групп острых волн с преимущественной локализацией в левой теменно-затылочной области. При магнитно-резонансном исследовании мозга выявлены ишемические очаги в области головки хвостатого ядра и в белом веществе головного мозга кнаружи от внутренней капсулы слева.
Изменения со стороны сердца характеризовались в одном случае симметричной гипертрофической кардиомиопатией (толщина межжелудочковой перегородки и толщина задней стенки левого желудочка составили 11мм при норме 7 мм) в сочетании с нарушением сердечного ритма (эктопический предсердный ритм, суправентрикулярная экстрасистолия). В другом случае наблюдалась дилатационная кардиомиопатия (конечно-диастолический диаметр левого желудочка составил 56 мм при норме 40 мм) со снижением контрактильной способности миокарда (фракция выброса составила 0,34 при норме 0,60). По данным биохимического анализа крови в том и другом случае отмечалось повышение содержания молочной (в исходе– 2,8 ммоль/л, через 1 час после нагрузки глюкозой – 3,0, через 3 часа – 3,2 ммоль/л, при норме1,0 - 1,7 ммоль/л) и пировиноградной кислоты (в исходе – 0,34 ммоль/л, через 1 час – 0,28, через 3 часа – 0,24 ммоль/л, при норме 0,05 - 0,09 ммоль/л). По данным биопсии скелетной мышцы в 35% мышечных волокон определялся выраженный феномен RRF, накопление микроконгломератов солей кальция.
Под нашим наблюдением находился 1 ребенок с синдромом MERRF. В основе этого синдрома лежит точковая мутация в позиции 8344 в гене лизиновой tРНК. Экстракардиальными симптомами, доминирующими в клинической картине являлись миоклонус-эпилепсия, атаксия, деменция, потеря слуха и мышечная слабость. Изменения со стороны сердца характеризовались симметричной гипертрофической кардиомиопатией (толщина межжелудочковой перегородки и толщина задней стенки левого желудочка составили 12мм, при норме 7 мм). Отмечался синдром Вольфа-Паркинсона-Уайта, что создавало предпосылки для возникновения жизнеугрожаемых состояний.
Синдром Барта, описан в 1983 г. P.G. Barth как Х-сцепленный рецессивный фенотип, проявляющийся сочетанием скелетной миопатии, кардиомиопатии, задержки роста и нейтропении в раннем возрасте. О митохондриальной природе заболевания свидетельствуют резко выраженные нарушения строения митохондрий мышечной, сердечной ткани и других органов [19]. Тяжесть заболевания, в первую очередь обусловлена степенью поражения сердечной мышцы, которая определяет прогноз состояния детей. Возможна как дилатационная так и гипертрофическая кардиомиопатии. Мы наблюдали мальчика с синдромом Барта, у которого отмечена симметричная необструктивная гипертрофическая кардиомиопатия (толщина межжелудочковой перегородки и толщина задней стенки левого желудочка составили 11мм, при норме 7 мм). К экстракардиальным симптомам относились: задержка физического развития (рост <3 центиля, масса <10 центиля), миопатический синдром, верифицируемый данными миографии, нейтропения. Отмечалась гипогликемия (2,0 – 3,12 ммоль/л, при норме 3,3 - 5,7 ммоль/л), повышение уровней молочной кислоты молочной кислоты (4,16 ммоль/л, при норме 1,0 - 1,7 ммоль/л) и пировиноградной кислот(0,17 - 0,23 ммоль/л при норме 0,05 - 0,09 ммоль/л), повышенная экскреция 3-метилглутаконовой, 3-метилглутаровой, 3-гидрокси-3-метилглутаровой кислот. Содержание общего карнитина в сыворотке было снижено (7,6 при норме 47,4 мкмоль/л). По данным биопсии скелетной мышцы обнаружены субсарколеммальные скопления митохондрий с признаками их деструкции, феномен «рваных красных волокон» отмечен в 15% волокон, выявлены скопления мелкозернистых конгломератов кальция.
Важнейшей специфической составляющей внутренней мембраны митохондрий являются ферментные комплексы дыхательной цепи, осуществляющие перенос электронов к молекуле кислорода. Выделяют 5 основных ферментных комплексов: I комплекс – НАДФ–коэнзим-Q-редуктаза; II комплекс – сукцинат–коэнзим-Q-редуктаза; III комплекс – коэнзим-Q–цитохром-С1-редуктаза; IV комплекс – цитохромоксидаза; V комплекс – АТФ-синтетаза. Патология одного или нескольких ферментных комплексов дыхательной цепи может быть причиной развития кардиомиопатий.
Дефицит I комплекса в клинической картине проявляется мышечной слабостью, умственной отсталостью, офтальмоплегией, лактат-ацидозом, дилатационной (от 3 до 5 лет) или гипертрофической (после 5 лет) кардиомиопатией. При биопсии мышцы отмечается феномен RRF, накопление липидов и паракристаллических включений в митохондриях.
Дефицит II комплекса характеризуется мышечной слабостью, вторичным дефицитом карнитина, лактат-ацидозом, дилатационной или гипертрофической кардиомиопатией.
Дефицит III комплекса проявляется как гистиоцитарная кардимиопатия. Девочки болеют чаще мальчиков (5:1). Клиническая симптоматика заболевания появляется, начиная с 3-х недельного возраста и, как правило, до 1 года. Характеризуется внезапным возникновением тахиаритмий желудочковой или наджелудочковой локализации, возможно развитие фибрилляции желудочков, приводящее к внезапной смерти. Нами наблюдалась девочка одного года двух месяцев с гистиоцитарной кардиомиопатией. Изменения со стороны сердца характеризовались дилатационной и гипертрофической симметричной необструктивной кардиомиопатией с фиброэластозом эндокарда, тяжелыми приступами пароксизмальной тахикардии на фоне синдрома Вольфа-Паркинсона-Уайта. Девочка погибла на фоне одного из приступов пароксизмальной тахикардии, осложненного фибрилляцией желудочков. При аутопсии выявлено сочетание гипертрофии миокарда, с дилатацией полости левого желудочка, фиброэластоз эндокарда. При световой микроскопии обнаружены необычные очаги «обесцвеченного» миокарда, главным образом в желудочках. Эти очаги содержали удлиненные мышечные волокна - так называемые гистиоцитоподобные, пенистые, или онкоцитарные клетки. В цитоплазме гистиоцитоидных клеток содержится большое количество липидов и гликогена. По данным электронной микроскопии в гистиоцитоидных клетках содержалось большое количество митохондрий причудливой формы.
Дефицит IV комплекса характеризуется гипертрофической кардиомиопатией, мышечной слабостью, почечной дисфункцией, лактат-ацидозом, вторичным дефицитом карнитина.
Кардиомиопатии могут возникать при дефиците митохондриальных ферментов, ответственных за метаболизм жирных кислот и пирувата. При этом развитие кардиомиопатий происходит на фоне первичного или вторичного дефицита карнитина. Нами наблюдались 2 больных с карнитиновой кардиомиопатией. Дефицит карнитина клинически характеризуется медленно прогрессирующей мышечной слабостью, изменения со стороны сердца характеризуются дилатационной или гипертрофической кардиомиопатией с развитием сердечной недостаточности на поздних стадиях заболевания, нарушениями проводящей системы сердца в виде патологической брадикардии. Приводим одно из наблюдений.
Больной А. 9-ти лет с карнитиновой недостаточностью, вызвавшей развитие дилатационной кардиомиопатии. В возрасте 7 лет появились жалобы на повышенную утомляемость, слабость, потливость, на частые боли в животе, выраженный миопатический синдром, признаки недостаточности кровообращения. В стационаре по месту жительства был поставлен диагноз: дилатационная кардиомиопатия. Назначенное лечение (дигоксин в комбинации с мочегонными препаратами) оказалось неэффективным нарастали явления недостаточности кровообращения, увеличивалась дилатация левого желудочка. В отделении кардиологии Московского НИИ педиатрии и детской хирургии Росздрава на основании экстракардиальных симптомов (гепатомегалия, миопатический синдром) в сочетании с особенностями изменений со стороны сердечно-сосудистой системы в виде сочетания дилатации левого желудочка (конечно диастолический диаметр левого желудочка составил 66 мм, при норме 41 мм), снижения контрактильной способности миокарда (фракция выброса=0,32) с явлениями гипертрофии стенок левого желудочка (толщина межжелудочковой перегородки и толщина задней стенки левого желудочка составили 12 мм, при норме 8 мм), усиления сигнала от эндокарда, и специфических изменений на ЭКГ в виде гигантских Т зубцов в левых грудных отведениях, превышающих зубцы R, была заподозрена карнитиновая кардиомиопатия. С целью верификации диагноза был определен уровень карнитина в плазме. Уровень карнитина плазмы был снижен до 11 мкмоль/л при норме 35 мкмоль/л. В связи с этим к стандартной терапии сердечной недостаточности (дигоксин, мочегонные препараты) был подключен элькар из расчета 75 мг/кг в сутки. На фоне данной терапии состояние значительно улучшилось, уменьшились явления недостаточности кровообращения до 1 степени, сократились размеры полости левого желудочка (конечно диастолический диаметр левого желудочка уменьшился с 66 до 52 мм), улучшилась контрактильная способность миокарда (фракция выброса выросла с 0,32 до 0,49).
Митохондриальная дисфункция может быть причиной развития не полиорганной патологии, а проявляться преимущественным поражением миокарда. В связи с этим, нами проведена оценка выраженности нарушений клеточной энергетики при изолированных кардиомиопатиях.
Группа обследованных больных с дилатационной кардиомиопатией в зависимости от указаний в анамнезе на перенесенный миокардит подразделена на две подгруппы: больные с постмиокардитической и идиопатической формами. На основании результатов клинического наблюдения и инструментального обследования, нами были разработаны критерии компенсации и декомпенсации функции миокарда у больных с дилатационными кардиомиопатиями.
Критериями миокардиальной недостаточности являлись недостаточность кровообращения IIБ-III степени, комбинированная перегрузка желудочков и предсердий, II-III степень митральной недостаточности, легочная гипертензия, выраженная дилатация левого желудочка (увеличение конечно диастолический диаметр левого желудочка более 60 мм), резкое снижение сократительной способности миокарда (фракция выброса менее 0,4), выраженные ишемические изменения в миокарде, явления кардиосклероза, нарушения ритма высоких градаций ( по классификации B.Lown)..
Сравнительный анализ частоты встречаемости экстракардиальных симптомов митохондриальной дисфункции в подгруппах с идиопатической и постмиокардитической дилатационными кардиомиопатиями показал, что отставание в физическом развитии (ниже пятого перцентиля), не связанное с развитием недостаточности кровообращения, достоверно чаще встречалось при идиопатической дилатационной кардиомиопатии (89% против 23% случаев в подгруппе с постмиокардитической формой). Миопатический синдром также доминировал при идиопатической дилатационной кардиомиопатии (88% случаев против 14%, соответственно).
Средние показатели уровня лактата и пирувата при дилатационной кардиомиопатии были сравнимы с таковыми у детей с первичными митохондриальными нарушениями и максимально выражены при идиопатической дилатационной кардиомиопатии и постмиокардитической форме в стадии декомпенсации. Исходная гипогликемия и транзиторная гипергликемия достоверно чаще встречались при идиопатической дилатационной кардиомиопатии.
В группу больных с гипертрофической кардиомиопатией вошли 14 пациентов с асимметричной обструктивной и 12 с симметричной необструктивной формами. Анализ течения заболевания и степени выраженности клинических симптомов в каждой группе позволил выделить две стадии развития болезни: компенсации и декомпенсации. Критериями стадии декомпенсации являлись жалобы на одышку, стенокардические боли в сердце, синкопальные состояния, признаки недостаточности кровообращения II степени, комбинированная перегрузка желудочков и предсердий, гипертрофия межжелудочковой перегородки более 20 мм, градиент внутрижелудочкового давления более 40 мм ртутного столба, выраженные ишемические изменения, нарушения ритма высоких градаций (по классификации B.Lown) и нарушение сердечной проводимости.
При асимметричной обструктивной гипертрофической кардиомиопатии преобладала стадия декомпенсации - 78% случаев. При симметричной гипертрофической кардиомиопатии заболевание длительно протекало бессимптомно, степень гипертрофии миокарда не превышала 15 мм, отсутствовал градиент обструкции, выраженность миокардиальной недостаточности соответствовала стадии компенсации. В то же время эти больные имели выраженные экстракардиальные симптомы митохондриальной патологии, такие как: инфантильный соматотип (показатели физического развития ниже 5 перцентиля), умеренный миопатический синдром, лактат-ацидоз, транзиторная гипогликемия, материнское наследование.
В одном наблюдении при несвоевременной коррекции нарушений клеточной энергетики в миокарде у больного с симметричной гипертрофической кардиомиопатией, возникшей на фоне митохондриальной дисфункции, в течение наблюдения произошел переход заболевания из стадии компенсации в стадию декомпенсации. При этом наряду с нарастанием гипертрофии миокарда развилась миогенная дилатация левого желудочка, снизилась сократительная способность миокарда, возникли признаки недостаточности кровообращения II степени.
При гипертрофической кардиомиопатии показатели уровней лактата и пирувата были сравнимы с группой больных с первичной митохондриальной патологией и максимально выражены при симметричной форме гипертрофической кардиомиопатии.
При анализе морфологических маркеров митохондриальной дисфункции по данным гистохимического анализа биопсии скелетной мышцы у всех обследованных с дилатационной кардиомиопатией и у 82% с больных с гипертрофической кардиомиопатией выявлены признаки митохондриальной патологии. По данным биопсии скелетной мышцы количество RRF у пациентов с дилатационной кардиомиопатией (25±4,1%) и гипертрофической кардиомиопатией (21,7±3,4%) было сопоставимо с количеством RRF при первичных митохондриальных нарушениях. Согласно современным представлениям наличие RRF является общепринятым маркером полисистемной митохондриальной недостаточности. В норме их количество не превышает 5%. Появление этих волокон может быть непосредственным следствием мутации митохондриальной ДНК или своеобразным компенсаторным эффектом в ответ на функциональную слабость митохондрий. Значения интегрального параметра митохондриальной недостаточности – митохондриального индекса, включающего как характеристику RRF, так и оценку степени активности митохондриальных ферментов (сукцинатдегидрогеназы и цитохромоксидазы) в скелетных мышцах, у детей с кардиомиопатиями в целом были сходны с таковыми у больных с митохондриальными синдромами. Митохондриальный индекс составил при дилатационной кардиомиопатии 2,05±0,09, при гипертрофической кардиомиопатии –1,92±0,11, при первичных митохондриальных нарушениях – 2,25±0,10 (норма – до 1,0). Кроме феномена RRF при световой микроскопии отмечены субсарколеммальные скопления гликогена, кальция и липидов, что является важным косвенным признаком митохондриальной недостаточности. При электронной микроскопии определялись аномальные полиморфные митохондрии. Аномалии касались количества, величины, формы, и внутренней структуры митохондрий (Рис.1.).
Вместе с тем выраженность морфологических маркеров митохондриальной патологии у обследованных больных была неоднородна, были выделены три степени выраженности митохондриальной дисфункции (таблица 1).
Была проанализирована степень выраженности митохондриальной дисфункции при различных клинических вариантах кардиомиопатий. Выраженные митохондриальные нарушения (митохондриальная дисфункция III степени) были выявлены при идиопатической дилатационной кардиомиопатии и симметричной гипертрофической кардиомиопатии. При этом морфологические признаки митохондриальной дисфункции сочетались с экстракардиальными симптомами митохондриальной недостаточности. Следует подчеркнуть, что у больных этих групп отсутствовали признаки недостаточности кровообращения.
Таблица № 1. Степень выраженности митохондриальной дисфункции по данным патоморфологических параметров биопсии скелетной мышцы
* См. объяснение в тексте
Признак I степень, слабо выраженная II степень, умеренно выраженная III степень, выраженная количество RRF* от 10 до 15% от 15 до 25% более 25% Митохондриальный индекс* от 1,25 до 1,5 от 1,5 до 2,25 более 2,25 Липиды Нет от 1 до 1,5 баллов более 1,5 баллов Кальций Нет от 1 до 2 баллов более 2 баллов Гликоген Нет от 1 до 1,5 баллов более 1,5 баллов Ультраструктурные изменения митохондрий не выражены частично выражены сильно выражены В группе с симметричной гипертрофической кардиомиопатией при морфологическом исследовании скелетной мышцы в большинстве случаев выявлялись нарушения функции митохондрий (наличие феномена RRF, изменение активности митохондриальных ферментов, ультраструктурные аномалии митохондрий, нарушение распределения кальция, липидов и гликогена в биоптате скелетной мышцы). Выраженность этих маркеров патологии соответствует митохондриальной недостаточности III степени и сопоставимы с таковыми при первичных митохондриальных синдромах.
Выявление сходства проявлений митохондриальной недостаточности при первичных митохондриальных синдромах, идиопатических дилатационных кардиомиопатиях и симметричной необструктивной форме гипертрофической кардиомиопатии позволяет предположить первичный характер митохондриальной дисфункции в этих группах.
Нами установлено, что при «постмиокардитической» дилатационной кардиомиопатии существует сопряженность между степенью нарушений функции митохондрий и выраженностью миокардиальной дисфункции. Миокардиальная функция в стадии компенсации сочетается со слабовыраженной митохондриальной дисфункцией. Миокардиальная функция в стадии декомпенсации, характеризующаяся ишемическими изменениями в миокарде, степенью недостаточностью кровообращения IIБ, сочетается с выраженными нарушениями функций митохондрий. Аналогичные изменения были выявлены в группе больных с асимметричной гипертрофической кардиомиопатией.
Таким образом, полученные данные позволяют заключить, что метаболические нарушения, связанные с митохондриальной дисфункцией, являются важным патогенетическим аспектом развития кардиомиопатий. Развитие метаболических изменений при кардиомиопатиях могут быть обусловлены как первичным, так и вторичным нарушением митохондриальных функций. Стадия декомпенсации при кардиомиопатиях, сопровождаемая выраженными ишемическими изменениями в миокарде, недостаточностью кровоснабжения IIБ степени сопряжена с вторичной митохондриальной недостаточностью гипоксического генеза.
В виду этого представляется необходимым своевременно диагностировать митохондриальные нарушения при кардиомиопатиях.
Нами разработаны диагностические критерии, позволяющие предположить развитие кардиомиопатий, на фоне первичной энергетической несостоятельностью митохондрий:
1. инфантильный соматотип (вес и рост ребенка соответствует 3-5 перцентилю, отставание костного возраста);
2. мышечная слабость, выявляемая в покое или после физической нагрузки;
3. периодическая (циклическая) нейтропения;
4. стойкое увеличение печени, не связанное с явлениями сердечной недостаточности на фоне приема сердечных гликозидов и диуретиков;
5. семейный характер заболевания, чаще материнское наследование;
6. врожденная катаракта;
7. птоз в сочетании с атриовентрикулярной блокадой;
8. кардиомегалия в сочетании с желудочковыми аритмиями у детей раннего возраста;
9. сочетание дилатации полостей сердца и гипертрофии миокарда, преимущественно задней стенки с проявлениями фиброэластоза эндокарда;
10. выявление симметричной гипертрофической кардиомиопатии в раннем возрасте;
11. "гигантские" зубцы Т на ЭКГ в левых грудных отведениях;
12. повышение уровня лактата более 2,5 ммоль/л и пирувата более 0,2 ммоль/л, увеличение соотношения лактат/пируват более 20, лактат-ацидоз;
13. энцефалопатия;
14. транзиторная гипогликемия.
Для верификации митохондриальной дисфункции следует использовать: данные морфологических параметров биопсии скелетной мышцы (выявление RRF, снижение активности сукцинатдегидрогеназы и цитохромоксидазы, наличие липидных включений, отложений липидов и кальция, аномалии ультраструктуры митохондрий);
Глубокие метаболические изменения в миокарде, в первую очередь нарушения механизмов энзимной регуляции энергетического обмена, служат основой для применения у больных с кардиомиопатиями препаратов, улучшающих метаболизм пораженного миокарда. Фармакологическая коррекция метаболических нарушений при кардиомиопатиях у детей направлена на улучшение разных звеньев энергетического обмена. Для этого используются кофакторы энергетических реакций энзимного обмена, препараты, осуществляющие перенос электронов в дыхательной цепи митохондрий.
L-карнитин регулирует процессы энергообразования в клетке, что необходимо для нормальной жизнедеятельности организма. Только после связывания с L-карнитином происходит метаболизм жирных кислот в митохондриях с образованием энергии. Дефицит карнитина способствует накоплению жирных кислот в виде ацил-КоА. Увеличение концентрации ацил-КоА в цитоплазме обладает протоксическим действием по отношению к мембране клетки, приводит к нарушению транспорта АТФ, способствует повышению содержания кальция в цитоплазме и значительному повреждению сократительного аппарата миокарда [20].
L-карнитин выпускается за рубежом в виде пищевой добавки, содержащей 250-500 мг (в капсуле). На лекарственном рынке в России с 2000 года появился новый препарат L-карнитина (Элькар®), представляющий собой 20% раствор левокарнитина, синтезируемый на основе природного L-карнитина.
Своевременная терапия L-карнитином дилатационной кардиомиопатии на фоне первичной или вторичной карнитиновой недостаточности позволяет избежать неблагоприятного течения заболевания и достичь выраженного благоприятного клинического эффекта. Показано, что назначение L-карнитина в дозе 50-100 мг/кг в сутки в течение 3-6 мес при карнитиновой кардиомиопатии позволяет нормализовать размеры сердца, компенсировать сердечную недостаточность [21]. Аналогичные результаты получены при наблюдении за 4-х месячным мальчиком с дилатационной кардиомиопатией, обусловленной дефицитом карнитина. Общий уровень карнитина в плазме составлял 25 мкмоль/мл. Применение L-карнитина способствовало нормализации функции левого желудочка [14].
M.E. Pierpont и соавт. [22] приводят наблюдение за тремя детьми с тяжелыми формами дилатационной кардиомиопатии, возникшей на фоне транспортных дефектов карнитинового обмена. Выявление значительного снижения уровня карнитина в плазме и скелетных мышцах послужило основанием для заместительной терапии L-карнитином. Данная терапия способствовала уменьшению выраженности дилатационной кардиомиопатии. Делается заключение, что своевременная терапия L-карнитином дилатационной кардиомиопатии, связанной с транспортными дефектами карнитина, позволяет избежать неблагоприятного течения заболевания и достичь выраженного клинического эффекта.
J. Draaisma с соавт. [23] приводит описание успешного лечения L-карнитином дилатационной Х-сцепленной кардиомиопатии, сходной по клинической картине с карнитиновой кардиомиопатией, обусловленной вторичной метилглютаконовой ацидурией II типа.
Мы обладаем успешным опытом лечения симметричной гипертрофической кардиомиопатии у мальчика с синдромом Барта и резко увеличенной экскрецией 3-метилглутаконовой, 3-метилглутаровой и 3-гидрокси-3-метилглутаровой кислот. На фоне терапии, включавшей L-карнитин 500 мг/сут (ЭлькарÒ), цитомак 8,0 (30 мг), 5 внутривенных инъекций, коэнзим Q10 60 мг/сут. была достигнута нормализация толщины межжелудочковой перегородки (с 11 до 7,5 мм) и задней стенки левого желудочка (с 11 до 7 мм). Улучшилось клиническое состояние мальчика (повысился мышечный тонус, увеличилась толерантность к физическим нагрузкам), отмечено снижением экскреции 3-метилглутаконовой, 3-метилглутаровой и 3-гидрокси-3-метилглутаровой кислот.
Важно отметить, что применение L-карнитина возможно не только при лечении митохондриальных кардиомиопатий, но и при лечении идиопатических кардиомиопатий, при которых нарушения клеточной энергетики носят вторичный характер, обусловленный развитием сердечной декомпенсации.
Сердечная декомпенсация сопряжена с нарушением клеточной энергетики и, в первую очередь, с патологией карнитинового метаболизма и тесно связанного с ним β-окисления жирных кислот, а также изменениями в цепи дыхательных ферментов митохондрий [3,20]. Нарушение метаболизма жирных кислот является важным звеном патогенеза недостаточности кровообращения, что проявляется внутриклеточной аккумуляцией жирных кислот, ацил-карнитина, ацил-КоА. Применение L-карнитина для лечения ишемического поражения миокарда основываются на его способности связывать излишки ацил-КоА, удалять жирные кислоты из цитоплазмы, и, таким образом, снижать токсические эффекты [24].
В исследовании I. Rizos [25] оценивалась возможность применения L-карнитина для лечения недостаточности кровообращения у взрослых больных с дилатационной кардиомиопатией. Обследовано 80 больных с дилатационной кардиомиопатией с недостаточностью кровообращения III – IV класса (функциональные классы Нью-Йоркской Сердечнойя Ассоциации). Оценивалась смертность в течение трехлетнего наблюдения. После 3-месячного периода стабилизации пациенты были рандомизированы на 2 группы. 1- группа получала L-карнитин (2 г в сутки), 2- группа плацебо. Группы статистически не отличались по клиническим и гемодинамическим параметрам в начале исследования. Проведенный статистический анализ выживаемости с использованием метода Kaplan-Meier выявил статистически достоверный эффект, улучшающий продолжительность жизни при применении L-карнитина. Делается вывод, что применение L-карнитина должно обязательно входить в комплексную терапию сердечной недостаточности у больных с дилатационной кардиомиопатией.
В ходе мультицентрового проспективного исследовании была оценена возможность применения L-карнитина для лечения кардиомиопатий у детей [26]: 76 пациентов с кардиомиопатией лечились L-карнитином в сочетании с традиционной терапией, контрольную группу составили 145 пациентов, принимавших только традиционную терапию лечения сердечной недостаточности (ингибиторы ангиотензинпревращающего фермента, мочегонные препараты). Длительность применения L-карнитина варьировала от 2 недель до 1 года и более. При этом оценивалась выживаемость, выраженность недостаточности кровообращения, динамика эхокардиографических параметров. В начале исследования пациенты, получавшие L-карнитин, были моложе, чем пациенты контрольной группы и имели худшие инструментальные и клинические параметры. В конце исследования в группе, получавшей L-карнитин, отмечены более низкие показатели смертности, лучшая динамика клинических и эхокардиографических показателей по сравнению с контрольной группой. Кроме того, было продемонстрировано, что изолированное применение ингибиторов ангиотензинпревращающего фермента приводило к большей смертности. Таким образом, в ходе этого исследование доказан благоприятный клинический эффект применения L-карнитина в комплексной терапии пациентов с кардиомиопатиями.
В исследовании S.Winter и соавт. [27] было продемонстрировано, что комбинация инотропных и мочегонных препаратов в сочетании с метаболической терапией L-карнитином способствует увеличению продолжительности жизни у детей с кардиомиопатиями.
Нами [28] было проведено сравнительное исследование по применению L-карнитина у детей с кардиомиопатиями. Дети были разделены на две подгруппы. Дети из 1-ой получали L-карнитин (использовался Элькар) в комплексе с традиционной терапией недостаточности кровообращения (дигоксином в сочетании с ингибиторами ангиотензинпревращающего фермента и мочегонными), из 2-ой только традиционную терапию. В исходе группы не отличались по выраженности степени недостаточности кровообращения и показателям контрактильной способности миокарда. Доза Элькара составляла 50 мг/кг массы тела в сутки. Установлено, что через 6 месяцев дети, получавшие Элькар, имели достоверно меньшую степень недостаточности кровообращения и более высокие показатели фракции выброса.
Выводы:
1. Проведенное исследование выявило, что нарушение клеточной энергетики, является клинико-патогенетической основой кардиомиопатий у детей. Митохондриальная дисфункция при идиопатической дилатационной и симметричной гипертрофической кардиомиопатиях сходна по своим проявлениям с первичными митохондриальными синдромами (Кернса-Сейра, MELAS, Барта, карнитиновые кардиомиопатии) и проявляется сочетанием кардиальных симптомов (кардиомегалия не связанная с перенесенным миокардитом, симметричный характер гипертрофии миокарда) с экстракардиальными симптомами митохондриальной патологии (инфантильный соматотип, миопатический синдром, гиперлактат- и гиперпируватемия).
2. Стадия декомпенсации при кардиомиопатиях, сопровождаемая выраженными ишемическими изменениями в миокарде, недостаточностью кровооснабжения II Б степени сопровождается вторичной митохондриальной недостаточностью гипоксического генеза.
3. Для генетических синдромов, возникающих на фоне первичной митохондриальной патологии, характерен клинический полиморфизм поражения сократительного миокарда по типу дилатационной или симметричной гипертрофической кардиомиопатии в сочетании с нарушениями в проводящей системы сердца с развитием жизнеугрожаемых тахи- или брадиаритмий.
4. План обследования ребенка с кардиомиопатиями с подозрением на митохондриальную патологию должен включать электрокардиографию, эхокардиографию, электроэнцефалографию, электромиографию, консультации невролога, окулиста, генетика, определение уровня лактата и пирувата на фоне стандартного глюкозотолерантного теста, уровня органических кислот (масс-спектрометрия мочи), сахарную кривую, оценку кислотно-щелочного состояния, биопсию скелетной мышцы (световая и электронная микроскопия).
5. Установлено, что митохондриальная дисфункция при кардиомиопатиях поддается терапевтической коррекции. Применение L-карнитина для лечения кардиомиопатий позволяет улучшить функциональное состояние миокарда, способствует устранению сердечной декомпенсации, снижает показатели смертности.
Литература
1. Braundwald Heart Disease A textbook of Cardiovascular Medcine 6th ed 2001: 1751-1792.
2. Амосова Е.Н. Кардиомиопатии. Киев: Книга плюс,999;421.
3. Кушаковский М.С. Хроническая застойная сердечная недостаточность, идиопатические кардиомиопатии.Cт-Петербург: Фолиант 1998;318. 4. Леонтьева И.В. Лекции по кардиологии детского возраста М.: Медпрактика 2005;138-275.
5. Garsia Marin, J. Goldenth M. Cardiomyopathies and anomal mitochondrial function. Cardiovascular Res 1994;28;4:456-463.
6. Sidi D., Le Bidois J,Pechaud J. . Enzymatic activities of mitochondrial respiratory chain child with cardiomyopathies 34 case prospectively studies by endomyocardial biopsy. Arch MollCocurVais 1992;.85: 5
7 stin P., Labidois J., Christien D. et.al. Endomyocardial biopsies for early detection of mitochondrial disorders in hyperthrophical cardiomyopathies. J. Pediatr 1994; 124: 2:224-228.
8. Ozava T. Mitochondrial cardiomyopathy. Herz 1994;19:2:105-118
9. Sato W., Tanaka M., Sugyama S. et.al. Cardiomyopathy and angiopathy in patients with mithochondrial myopathy encephalopathy lactic acidosis and stroke like episodes. Amer Heart J 1994;128: 4:733-741.
10. Arpa-J, Campos-Y, Cruz-Martinez-A, et al. Clinical and investigative approaches in mitochondrial disease. A review of 15 cases. Neurologia 1994; 9:8:324-336.
11. Molnar-M; Neudecker-S; Schroder-JM - Increase of mitochondria in vasa nervorum of cases with mitochondrial myopathy, Kearns-Sayre syndrome, progressive external ophthalmoplegia and MELAS. Neuropathol-Appl-Neurobiol 1995; 21:: 432-437
12. Леонтьева И.В., Николаева Е.А., Сухоруков В.С Варианты поражения сердечно-сосудистой системы при синдроме Кернса Сейра. Рос вест перинатол и педиатр 1999;6,26-32.
13. Angelini C., Melacini P., Valente M. et.al. Hypertrophic cardiomiopathy with mitochondrial myopathy (A new phenotype of complex II defects) Jpn.Heart J.;1993;34: 1:63-77.
14. .Zales V.R.; Benson D.W. Jr Reversible cardiomyopathy due to carnitine deficiency from renal tubular wasting. Pediatric-Cardiology 1995; 16:2:76-78.
15. Белозеров Ю.М. Недостаточность карнитина у детей. Рос вест перинатол и педиатр 1996;4,42-47.
16. Zeviani M., Mariotti C., Antozzi C., Fratta GM., Rustin P., Prelle A. Oxphos defects and mitochondrial DNA mutations in cardiomyopathy Muscle-and-Nerve 1995; 18/SUPPL.: 3,170-174.
17. Aoyama T., Souri M., Ushikubo-S., Kamijo T., Yamaguchi S. Purification of human very-long-chain acyl-coenzyme A dehydrogenase and characterization of its deficiency in even patients. J Clin Invest 1995; 95/6:2465-2473.
18. Consalvo D, Villegas F, Villa A.M, Kohler G. Severe cardiac failure in Kearns-Sayre syndrome Medicina, 1997,57,(1),67-71.
19. Николаева Е.А., Леонтьева И.В., Сухоруков В.С. Митохондриальный синдом Барта, благоприятное течение на фоне комплексной терапии. Вестник перинатологии и педиатрии 1998;43:5:37-40.
20. Леонтьева И.В. Роль L-карнитина в метаболизме миокарда и возможности его применения для лечения заболеваний сердца. Научный обзор, М 2002;31.
21. Белозеров Ю.М. Кардиологические аспекты патологии митохондрий. «Актуальные вопросы кардиологии детского возраста. М 1992;38.
22. Pierpont M.E; Judd D., Goldenberg I. Myocardial carnitine in end-stage congestive heart failure. Am J Cardiol 1989;64:56-80.
23. Draaisma J.T., Van-Kesteren I.C., Daniels O., Sengers R.C. Dilated cardiomyopathy with 3-methylglutaconic aciduria. Pediatr-cardiol 1994; 15/2: 89-90.
24. Порядин Г.В. Молекулярные механизмы повреждения клеток. Методические разработки. М: 1997;49.
25. Rizzon P; Iliceto S L-carnitine in the treatment of left ventricular dysfunction in post-infarction Cardiologia 1995; 12: 40: 12: Suppl 1: 41-3
26. Helton E., Darragh R., Francis P., Fricker F.J., Jue K., Koch G., Mair D., Pierpont M.E., Prochazka J.V., Linn L.S., Winter S.C. Metabolic aspects of myocardial disease and a role for L-carnitine in the treatment of childhood cardiomyopathy. Pediatrics 2000; 06: 105: 6: 1260-70.
27. Winter S.C., Buist N.R. Cardiomyopathy in childhood, mitochondrial dysfunction, and the role of L-carnitine. Am Heart J,2000 02; 139: 2 Pt 3, S63-9.
28. Себелева И.А. Митохондриальная дисфункция при кардиомиопатиях у детей и обоснование путей ее коррекции. Автореферат дисс. канд. мед. наук. М 2000; 27.
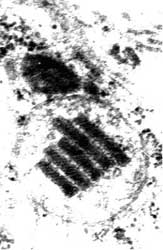
А. Крупная митохондрия с деструкцией большинства крист и наличием кристаллоидного включения. Электронная микроскопия, увеличение 110.000. Больной Б.Щ., 14 лет Диагноз: синдром Кернса-Сейра (препарат А.И. Клембовского).
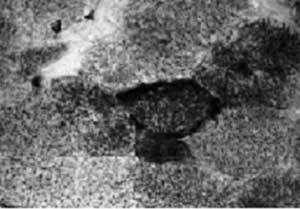
Б. Выраженный феномен RRF (гистохимическое выявление сукцинатдегидрогеназы). Световая микроскопия, увеличение 400. Больная И., 13 лет. Диагноз: синдром MELAS, дилатационная кардиомиопатия.
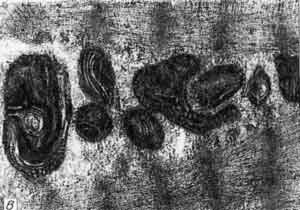
В. Полиморфные аномальные митохондрии среди миофибрилл. Различное расположение крист, их частичное слияние. Электронная микроскопия, увеличение 2200. Больной Ш,15 лет Диагноз: гипертрофическая кардиомиопатия.
Г
Г. Значительное повышение количества неодинаковых по размеру и форме митохондрий под сарколеммой мышечного волокна (участок RRF). Электронная микроскопия, увеличение 2200. Больная О., 10 лет. Диагноз: гипертрофическая кардиомиопатия.
Леонтьева Ирина Викторовна, доктор медицинских наук, профессор, главный научный сотрудник отделения кардиологии Московского НИИ педиатрии и детской хирургии Росздрава
Адрес для корреспонденции: 125412 Талдомская ул. д. 2
Телефон: +7(495) 483-71-01
Элькар -
| Октябрь 2007 г. |