Опубликовано в:
Педиатрическая фармакология 2003 №1(4)Коррекция недостаточности карнитина у детей с наследственными заболеваниями обмена веществ
Е.А.Николаева, А.Н.Семячкина, Е.С.Воздвиженская, М.Н.Харабадзе, П.В.Новиков
Московский НИИ педиатрии и детской хирургии Министерства здравоохранения РФПредставлен анализ собственных данных и сведений литературы о формах и причинах недостаточности карнитина у детей. Выявлен низкий уровень общего карнитина в крови у 16 пациентов с наследственной патологией обмена веществ, в том числе впервые установлена недостаточность карнитина у детей с синдромом Марфана и аутоиммунным полиэндокринным синдромом I типа. Продемонстрирована эффективность использования Элькара® для коррекции дефицита карнитина у детей.
Ключевые слова: карнитин, митохондриальные болезни, органические ацидемии, синдром Марфана, аутоиммунный полиэндокринный синдром I типа
Карнитин - биологически активное витаминоподобное вещество - был обнаружен в организме человека в начале ХХ века. Метаболическая роль карнитина была достаточно полно расшифрована лишь в течение последних десятилетий. Основными его функциями являются участие в энергетическом обмене, связывание и выведение из организма токсичных производных органических кислот.
Важная роль карнитина в биоэнергетических процессах заключается в том, что он принимает непосредственное участие в катаболизме липидов. Карнитинзависимые ферменты - ацилкарнитин-карнитинтранслоказа, карнитинпальми-тоилтрансферазы I и II - обеспечивают перенос длинноцепо-чечных жирных кислот в виде сложных эфиров (ацилкарни-тинов) из цитоплазмы через наружную и внутреннюю мито-хондриальную мембрану в матрикс митохондрий [1, 2]. Внутри митохондрий жирные кислоты подвергаются β-окислению с образованием ацетил-КоА, который служит субстратом для цикла трикарбоновых кислот Кребса и последующего синтеза АТФ в организме. Наряду с этим окисление жирных кислот - главный путь кетогенеза, а кетоновые тела являются дополнительным энергетическим источником для периферических тканей и головного мозга.
Влияние карнитина на жировой обмен осуществляется также его участием в цитоплазматическом синтезе жирных кислот путем обратного переноса необходимых для этого ацетильных групп митохондриального ацетил-КоА через ми-тохондриальную мембрану в цитоплазму [3].
Значение для организма карнитинзависимых процессов транспорта и окисления жирных кислот становится очевидным в условиях высокого расходования энергетических ресурсов, требующих повышенного катаболизма, то есть при интеркуррентных заболеваниях, усиленных физических или эмоциональных нагрузках, а также при недостаточном питании. После истощения запасов углеводов липиды становятся главным источником синтеза АТФ в организме [2, 4].
Помимо перечисленного, карнитин регулирует отношение ацил-КоА/свободный КоА в митохондриях. Путем связывания ацильного радикала он высвобождает КоА и тем самым активирует интенсивность энергетического метаболизма [5, 6].
Следующая важная функция карнитина заключается в его способности образовывать соединения с различными органическими кислотами, являющимися промежуточными
продуктами окислительных процессов. Данные вещества, которые накапливаются в митохондриях, цитоплазме клеток и в сыворотке крови, оказывают мембранотоксическое действие и ингибируют активность ряда ферментов. Выведение этих токсичных органических соединений из организма происходит через почки в виде ацилкарнитинов [7].
Эндогенное образование карнитина осуществляется в основном клетками печени и почек путем трансформации лизина; донатором метильных групп служит метионин [8]. Источником аминокислот служат белки пищевых продуктов, а также собственные протеины мышечной ткани. Однако эндогенный синтез обеспечивает суточную потребность в кар-нитине лишь приблизительно на 25%. Остальное количество должно поступать с пищей животного происхождения: мясом, рыбой, птицей и молочными продуктами; грудное молоко также содержит карнитин. Подсчитано, что потребность в карнитине у взрослых составляет от 200 до 500 мг/сут, при этом обычное меню обеспечивает поступление около 100-300 мг/сут [9].
В организме человека основное количество карнитина содержится в мышцах и сердце, что обусловлено высокой активностью липидного обмена в данных тканях. В связи с этим главными органами-«мишенями» при недостаточности карнитина служат скелетные мышцы и миокард, во вторую очередь страдают клетки головного мозга, печень и почки. Основные признаки дефицита карнитина: быстрая утомляемость, сниженная работоспособность, мышечная слабость, гипотония и гипотрофия, отставание физического и психомоторного развития, снижение школьной успеваемости, сонливость или раздражительность, нарушение функции сердца и печени, частые инфекционные заболевания - являются следствием развивающихся нарушений энергетического обмена и метаболизма липидов и связанных с ними расстройств других видов обмена веществ.
Благодаря наличию в своей структуре асимметрического атома углерода, молекула карнитина может существовать в двух изомерных формах: D- или L-стереоизомеров. В природных источниках обнаружен L-карнитин, и только он биологически эффективен. На важность понимания существенных различий между природным L-карнитином, D-карнити-ном и рацемическим D,L-карнитином (карнитина хлорид) было указано в ряде экспериментальных и обзорных работ. На основании многочисленных исследований было сделано два важных вывода:
1) только L-карнитин проявляет биологическую активность
2) D-карнитин негативно влияет на различные стороны метаболизма.
Так, установлено, что D-карнитин препятствует окислению жирных кислот в митохондриях и образованию энергии, конкурентно ингибирует активное усвоение L-карнитина органами и тканями, а также вызывает нарушение биосинтеза L-карнитина в печени, что приводит к его недостаточности в организме, особенно, в скелетных и сердечной мышцах, вызывая миастению и сердечную аритмию. Следовательно, D-карнитин представляет собой не просто неэффективное или биологически инертное вещество, а скорее вредную и потенциально опасную примесь. Именно поэтому уже в середине 80-х годов перестали применять рацемический карнитина хлорид, и в настоящее время во всем мире используются только препараты на основе L-карнитина.
Выделяют первичный и вторичный дефицит карнитина [7, 10].
Первичный дефицит, обусловленный дефектом транспорта карнитина в клетки и ткани, является генетическим детерминированным заболеванием с аутосомно-ре-цессивным типом наследования [2, 11]. Системная форма характеризуется резкой мышечной слабостью и гипотонией, тяжелой кардиомиопатией, жировой дистрофией печени и почек. При мышечной форме у больных наблюдается миопа-тия с накоплением липидов.
Причины вторичного дефицита карнитина более многообразны и встречаются гораздо чаще. Вторичная недостаточность характерна для большой группы наследственных заболеваний обмена веществ, в том числе для органических ацидемий, болезней транспорта и окисления жирных кислот. При этих состояниях низкий уровень карнитина в крови и тканях, главным образом, обусловлен активным выведением с мочой конъюгатов карнитина с токсичными органическими кислотами. Для митохондриальных болезней (синдрома Кернса-Сейра, прогрессирующей офтальмоплегии и др.) свойственен умеренный дефицит карнитина - повышенная потребность в нем связана с тяжелыми расстройствами электронного транспорта и окислительного фосфо-рилирования [12, 13].
Низкое содержание карнитина в тканях у детей с синдромом Ретта, повидимому, обусловлено нарушением процессов биоэнергетики, что проявляется повышенным уровнем лактата и пирувата в крови, активацией перекисного окисления липидов [14, 15]. При синдроме Рейе, манифестирующем на фоне вирусной инфекции, и у больных с эпилептическими синдромами на фоне лечения препаратами вальпроевой кислоты недостаточность карнитина, вероятно, вызвана расстройством метаболизма жирных кислот, высокой почечной экскрецией накапливающихся ацилкарнитинов [16]. Резко выраженный дефицит карнитина характерен для многих форм заболеваний сердца: гипертрофической, дилатационной, гистиоцитарной, диабетической кардиомиопатии, фиброэластоза эндокарда и др., что связано с высокой зависимостью миокардиальных энергетических процессов от липидного метаболизма [9].
Дети раннего возраста особенно чувствительны к недостаточности карнитина. Эндогенные запасы у них быстро истощаются при различных стрессовых ситуациях (инфекционные заболевания, желудочно-кишечные расстройства, нарушения вскармливания). Биосинтез карнитина резко ограничен в связи с небольшой мышечной массой, а поступление с обычными пищевыми продуктами не способно поддержать достаточный уровень в крови и тканях. Недоношенные и дети с малой массой особенно зависят от дополнительного получения карнитина [17].
У детей, страдающих расстройствами питания, рахитом, различными гастроинтестинальными заболеваниями (в том числе целиакией, муковисцидозом и др.) и болезнями почек, отмечается нарушение усвоения карнитина и его потеря через желудочно-кишечный тракт и почечные канальцы. Недостаточность карнитина отмечена у некоторых категорий пациентов, получающих специализированную диетотерапию, ограничивающую прием белковых продуктов, особенно животного происхождения (например, у больных с фенилкетонурией), а также у лиц, придерживающихся вегетарианской диеты [18].
Пациенты и методы
В отделе наследственных и врожденных заболеваний Московского НИИ педиатрии и детской хирургии МЗ РФ было обследовано 16 детей, страдающих различными формами наследственной патологии: в том числе митохондриальными синдромами Кернса-Сейра (3 ребенка) и Барта (1), нарушением окисления жирных кислот (4), изовалериановой ациде-мией (1), синдромом Марфана (4) и аутоиммунным полиэндокринным синдромом I типа (3). Возраст больных колебался от 3 до 14 лет.
Комплекс обследования детей включал анализ генеалогических данных, клинические и лабораторные методы, в том числе определение уровня общего карнитина в крови энзиматическим методом (с использованием наборов фирмы Roche, Германия), содержания молочной и пировино-градной кислот в крови, почечной экскреции органических кислот методом газовой хроматографии - хроматомасс-спектрометрии (Государственный антидопинговый центр). Диагноз у больных с синдромом Кернса-Сейра и аутоиммунным полиэндокринным синдромом I типа был подтвержден выявлением делеции митохондриальной ДНК и ядерной генной мутации в локусе 21q22.3 соответственно (Медико-генетический научный центр РАМН).
Результаты исследования и их обсуждение
Результаты проведенного обследования позволили выявить у детей комплекс клинических и лабораторных признаков, которые могли быть обусловлены имеющейся кар-нитиновой недостаточностью. В клиническом статусе у всех пациентов обращала внимание низкая толерантность к физической нагрузке, мышечная слабость и гипотония. У большинства больных отмечались снижение умственной работоспособности, головная боль, нарушение внимания и памяти, поражение сердца (в том числе кардиомиопатия), координа-торные и эндокринные расстройства. У половины пациентов было выявлено отставание физического развития, повторные приступы тошноты и рвоты, нарушение зрения (в том числе пигментный ретинит). У отдельных больных наблюдалось увеличение размеров и нарушение функции печени, тугоухость. Повышенный уровень лактата в крови (2,1-4,4 ммоль/л, норма 1,0-1,7 ммоль/л) был установлен у всех детей.
Уровень карнитина в крови оказался низким у всех обследованных больных. Как представлено в таблице, содержание общего карнитина в крови у детей составляло от 1,6 до 41,1 мкмоль/л, в среднем - 15,8 ± 3,6 мкмоль/л. Для контроля была использована сыворотка крови 10 детей, проходивших обследование в клинике и не имевших клинических признаков недостаточности карнитина. У них уровень карнитина колебался от 41 до 148 мкмоль/л и в среднем был равен 72,8 ±11,7 мкмоль/л.
Таблица. Уровень общего карнитина в крови у больных с генетически детерминированной патологией обмена веществ (n = 16)
Нозологическая форма
Содержание карнитина, мкмоль/л Синдром Кернса-Сейра (n = 3) 1,6; 32,9; 41,1 Синдром Барта (n = 1) 33,6 Нарушение окисления жирных кислот (n = 4) 14,5; 15,6; 27,3; 37,4 Изовалериановая ацидемия (n = 1) 4,1 Синдром Марфана (n = 4) 2,4; 8,2; 8,2; 24,7 Аутоиммунный полиэндокринный синдром Iтипа(n = 3) 4,1; 5,1; 16,4 Контроль (n = 10) 41,2-148,9 (72,8 ±11,7)
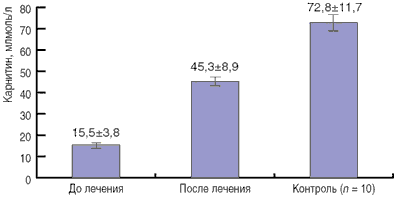
Рисунок. Динамика уровня общего карнитина в крови у больных с генетически детерминированной патологией обмена веществ через 4 нед после начала приема Элькара (n = 10).
Таким образом, у всех 16 пациентов с наследственной патологией содержание общего карнитина в крови было низким, хотя степень дефицита оказалась различной. Так, у 1 из 3 больных с митохондриальным синдромом Кернса-Сейра уровень карнитина в крови был лишь немного ниже границы нормы, а у другого ребенка с тем же заболеванием содержание карнитина было резко снижено (см. таблицу).
Для коррекции дефицита карнитина у наблюдавшихся больных был использован отечественный препарат Элькар (ООО «ПИК-ФАРМА», Россия), представляющий собой 20% водный раствор L-карнитина для перорального применения и содержащий 200 мг действующего вещества в 1 мл. Элькар производится из природного L-карнитина. Известно, что в тканях человека присутствует только L-стереоизомер, и именно он биологически эффективен. Дозы препарата составляли 20-30 мг на 1 кг массы тела в сутки. Однако ребенку с изовалериановой ацидемией была назначена более высокая доза - 50 мг/кг. У всех больных Элькар был применен в дополнение к основному лечению, и лишь при болезни Марфана данный препарат был использован в качестве монотерапии.
Помимо оценки клинического статуса больных у 10 детей через 4 нед после назначения Элькара был осуществлен контроль за изменением уровня карнитина в крови (см. рисунок), который показал достоверное его увеличение (в среднем в 3 раза). Среднее содержание общего карнитина в сыворотке крови у больных превысило нижнюю границу нормы, а у отдельных пациентов приблизилось к параметрам контрольной группы.
Увеличение уровня карнитина в крови сочеталось с улучшением состояния и самочувствия детей, снижением утомляемости, уменьшением выраженности миопатического синдрома, улучшением походки и координации движений, прекращением приступов метаболической декомпенсации.
Результаты обследования детей с болезнями обмена жирных кислот, органической ацидемией и митохондриальной патологией, таким образом, подтвердили имеющиеся в литературе сведения о недостаточности карнитина при перечисленных заболеваниях.
Обращали внимание впервые полученные данные о низком уровне общего карнитина в крови у 4 детей с моногенным заболеванием соединительной ткани - синдромом Марфана и у 3 больных с аутосомно-рецессивным аутоиммунным полиэндокринным синдромом I типа. Исследования, проведенные в нашей клинике, впервые позволили выявить у детей с синдромом Марфана митохондриаль-ную дисфункцию, проявляющуюся нарушением перекис-ного окисления липидов, нестабильностью цитомембран, угнетением процессов биологического окисления. На этом основании было высказано предположение о значении нарушений процессов биоэнергетики в патогенезе данной патологии [19]. Результаты настоящего исследования, свидетельствующие о недостаточности карнитина при синдроме Марфана, обосновывают необходимость включения карнитинсодержащих препаратов в комплекс лечения больных.
Результаты клинического обследования детей с аутоиммунным полиэндокринным синдромом I типа, наличие у них резко выраженной утомляемости, миопатических проявлений, мультисистемный характер поражения с вовлечением эндокринной системы, повышенный уровень молочной кислоты в крови заставили проводить дифференциальный диагноз с митохондриальной патологией. Однако у всех 3 больных окончательный диагноз аутоиммунного полиэндокринного синдрома был доказан обнаружением характерной для него генной мутации в локусе 21q22.3.
Таким образом, проведенные исследования доказали наличие недостаточности карнитина при ряде форм наследственных метаболических заболеваний и продемонстрировали высокую эффективность отечественного медикаментозного средства Элькар для коррекции дефицита карнитина. Несомненным достоинством препарата является простота применения, практическое отсутствие противопоказаний и побочных действий. Полученные результаты свидетельствуют, однако, что при большинстве форм генетически детерминированной патологии обмена веществ ликвидация карнитиновой недостаточности требует более длительного применения препарата и наблюдения за больными.
Литература
1.Eaton S., Bartlett K., Pourfarzam M. Mammalian mitochondrial β-oxidation. Biochem J 1996; 320: 345-57.
2. Brivet M., Boutron A., Slama A., et al. Defects in activation and transport of fatty acids. J Inherit Metab Dis 1999; 22: 4: 428-41.
3. Ленинджер А. Биохимия. Молекулярные основы структуры и функции клетки. Пер. с англ. М: Мир 1974.
4. Bartlett K., Pourfarzam M. Inherited disorders of mitochondrial fatty acid oxidation. Curr Pediatr 1997; 7:118-22.
5. Белозеров Ю.М. Недостаточность карнитина у детей. Российский вестник пе-ринатол. педиатр. 1996; 4: 42-7.
6. Shapira Y. Clinical aspects of mitochondrial encephalomyopathies. Int Pediatr 1993; 8: 225-32.
7. Stanley C.A. New genetic defects in mitochondrial fatty acid oxidation and carni-tine deficiency. Adv Pediatr 1987; 34: 59-88.
8. Var F.M., Ofman R., Fouchier S.W., et al. New insights in the enzymology and metabolism of carnitine byosynthesis in humans. J Inherit Metab Dis 2002; 25(7): 77.
9. Леонтьева И.В. Роль L-карнитина в метаболизме миокарда и возможности его применения для лечения заболеваний сердца. Научный обзор. М., 2002.
10. Stanley C.A., Berry G.T., Bennett M.J., et al. Renal handling of carnitine in secondary carnitine deficiency disorders. Pediatr Res 1993; 34: 89-97.
11. Christensen E., Vikre-Jorgensen J. Six years' experience with carnitine supplementation in a patient with an inherited defective carnitine transport system. J Inherit Metab Dis 1995; 18: 233-6.
12. Mandel H., Africk D., Blitzer M., Shapira E. The importance of recognizing secondary carnitine deficiency in organic acidemias: case report in glutaric academia type II. J Inherit Metab Dis 1988; 11:4: 397-402.
13. Hsu C.C., Chuang Y.H., Tsai J.L., et al. CPEO and carnitine deficiency overlapping in MELAS syndrome. Acta Neurol Scand 1995; 92: 252-5.
14. Ellaway C., Williams K., Leonard H., et al. Rett syndrome: randomized controlled trial of L-carnitine. J Child Neurol 1999; 14: 3:162-9.
15. Харабадзе М.Н., Улас В.Ю., Добрынина Э.В. и др. Фармакологическая коррекция митохондриальных нарушений при синдроме Ретта у детей. Педиатрическая фармакология 2003; 1: 45-9.
16. Hug G., McGraw C., Bates S., et al. Reduction of serum carnitine concentrations during anticonvulsant therapy with phenobarbital, valproic acid, phenytoin and carbamazepine in children. J Pediatrics 1991; 119: 5:799-802.
17. Кешишян Е.С., Казанцева Л.З., Николаева Е.А., Тозлиян Е.В. Использование препарата Элькар (L-карнитин) в педиатрии. Terra Medica 2001; 4: 42-3.
18. VilasekaM.A., Briones P., Ferrer I., et al. Controlled diet in phenylketonuria may cause serum carnitine deficiency. J Inherit Metab Dis 1993; 16:1:101-4.
19. Семячкина А.Н., Николаева Е.А., Семячкина С.В. и др. Медикаментозная коррекция нарушений клеточной биоэнергетики у больных с моногенными заболеваниями соединительной ткани (синдромы Марфана и Элерса-Данлоса). Педиатрическая фармакология 2003; 1: 41-4.Элькар -
| Октябрь 2007 г. |