Опубликовано в журнале:
«Русский журнал детской неврологии», ТОМ IV, выпуск 1, 2009, с. 3-10Леветирацетам в лечении юношеской миоклонической эпилепсии (предварительные результаты)
К.Ю. Мухин, М. Д. Тысячина, А.С. Петрухин
Кафедра неврологии и нейрохирургии ГОУ ВПО РГМУ Росздрава; Центр детской неврологии и эпилепсии, МоскваLevetiracetam In Treatment Of Juvenile Myoclonic Epilepsy (Preliminary Results)
K.Yu. Mukhin, M.D. Tysyachina, A.S. Petrukhin
Целью настоящего исследования было изучение эффективности и переносимости леветирацетама (кеппра, UCB) у больных юношеской миоклонической эпилепсией (ЮМЭ). Нами обследовано 12 пациентов с установленным диагнозом ЮМЭ в возрасте 14–22 лет, 4 мужчин и 8 женщин. Кеппра назначалась в комбинации с другими АЭП (вальпроаты, суксилеп) в 3 случаях и при монотерапии в 9 случаях (из них в 3 — в стартовой монотерапии). Катамнез составил от 7 мес. до 3 лет. Стойкое купирование эпилептических приступов было констатировано в 100% случаев, полное блокирование или выраженное уменьшение индекса интериктальных эпилептиформных разрядов на ЭЭГ — в 75%. У 5 из 6 пациентов кеппра достоверно уменьшила проявления фотосенситивности, как по данным клинической картины, так и по результатам ЭЭГ. Влияние кеппры на эпилептиформную активность было менее выражено в группе больных, получавших политерапию (комбинацию с вальпроатами или суксилепом) и при наличии абсансов (2 пациента). Побочные эффекты терапии кеппрой были отмечены лишь у одной пациентки (8%) в виде феномена насильственной нормализации Ландольта.
Ключевые слова: эпилепсия, юношеская миоклоническая эпилепсия, лечение, леветирацетам.
The aim of the study was to investigate the efficacy and tolerance of levetiracetam (keppra, UCB) in patients with juvenile myoclonic epilepsy (JME). Twelve patients diagnosed with JME, aged 14 to 22, were examined, including 4 males and 8 females. Keppra was combined with other anti-epileptic drugs (valproates, suxilep) in 3 cases, while in 9 cases it was administered as monotherapy (including initial monotherapy in 3 cases). The follow-up period varied from 7 months to 3 years. A stable relief of epileptic seizures was acknowledged in 100% cases; a complete blocking or an expressed reduction of the interictal epileptiform discharge index in EEG was achieved in 75% of patients. Keppra definitely reduced manifestations of photosensitivity in 5 out of 6 patients, which was proved both by the clinical presentation and EEG data. The impact of keppra on epileptiform activity was less expressed in the group of patients receiving polytherapy (combined with valproates or suxilep) and in cases with absences (2 patients). Side effects of keppra therapy were observed only in 1 female patient (8%), in the form of the Landolt’s forced normalization phenomenon.
Key words: epilepsy, juvenile myoclonic epilepsy, treatment, levetiracetam.
Юношеская миоклоническая эпилепсия (ЮМЭ) или синдром Янца — форма идиопатической генерализованной эпилепсии, характеризующаяся дебютом в подростковом возрасте с появлением массивных миоклонических приступов, возникающих преимущественно в период после пробуждения пациентов [3, 5]. В современной литературе заболевание впервые было описано D. Janz и W. Christian в 1957 году под названием «impulsive petit mal». С тех пор большое количество публикаций по данному синдрому было опубликовано в нашей стране и за рубежом, однако, установление точного диагноза все еще вызывает большие затруднения [3, 5, 20, 21, 28]. Основная ошибка врачей — поверхностно собранный анамнез, акцентирование внимания в истории болезни на генерализованных судорожных приступах (ГСП) и упускание из виду миоклонических приступов (МП) [2, 5, 22]. Panayiotopoulos С.P. и соавт. (1991) в Лондоне провели специальное статистическое исследование ошибок в диагностике ЮМЭ [22]. Авторы отметили, что из 70 обследованных ими больных у 66 (91,4%!) правильный диагноз установлен не был. Причем 1/3 из этих больных неоднократно проходили обследование и лечение в ведущих неврологических клиниках Великобритании. Согласно наблюдениям авторов, ЮМЭ правильно диагностировалась спустя лишь, в среднем, 8,3 лет с момента начала заболевания и спустя 17,7 мес. с момента посещения специализированной неврологической клиники. Вместе с тем, ЮМЭ — очень частая форма эпилепсии и, вероятно, одна из самых частых в рамках генерализованных эпилепсий. Частота ее среди всех форм эпилепсии составляет 5–12%, а среди идиопатических генерализованных форм — до 23% [28].
Дебют ЮМЭ варьирует от 7 до 21 года с максимумом в возрастном интервале 11–15лет. МП — облигатный тип приступов при данном заболевании. Миоклонические пароксизмы характеризуются молниеносными подергиваниями различных групп мышц; они чаще двухсторонние, симметричные, единичные или множественные, меняющиеся по амплитуде; нередко возникающие в виде серии залпов. Локализуются миоклонии, главным образом, в плечевом поясе и руках, преимущественно, в разгибательных группах мышц. У 30% пациентов миоклонические приступы захватывают мышцы ног, при этом больной ощущает внезапный удар под колени и слегка приседает или падает (миоклонически-астатические приступы) [5]. Сознание во время миоклонических приступов сохранено. Миоклонические приступы возникают или учащаются в первые минуты и часы после пробуждения пациентов. Снижение уровня бодрствования, сонливость, зевота, закрывание глаз — эти факторы нередко повышают вероятность появления приступов в утренние часы. В 90% случаев миоклонические приступы сочетаются с ГСП пробуждения. Генерализованному судорожному приступу может предшествовать серия миоклонических пароксизмов. Данный тип приступа называется клонико-тонико-клоническим. У 40% пациентов присоединяются короткие абсансы ювенильного (непикнолептического) типа [5]. Важнейшими факторами, провоцирующими приступы при ЮМЭ, являются депривация сна и внезапное насильственное пробуждение. У некоторых пациентов миоклонические приступы возникают исключительно при недосыпании. Примерно у 1/3 больных ЮМЭ (чаще женского пола) выявляется фотосенситивность. Возможно учащение ГСП и миоклонических приступов в перименструальном периоде [3, 5]. При неврологическом осмотре больных патологические изменения отсутствуют, когнитивные нарушения не характерны. У отдельных пациентов констатируется высокий уровень тревоги и невротизации, склонность к депрессивным реакциям [5]. Характерный ЭЭГ паттерн у больных ЮМЭ — короткие разряды генерализованной быстрой пик/полипик-волновой активности, иногда с некоторым опережением в лобных отведениях (рис. 1). Он провоцируется ритмической фотостимуляцией и закрыванием глаз. Эпилептиформная активность на ЭЭГ выявляется у 80–95% больных в межприступном периоде. Основная активность фоновой записи всегда сохранна. Изменения при нейровизуализации отсутствуют [5, 21].
Рис. 1. Пациентка Г.А., 17 лет. Диагноз: Юношеская миоклоническая эпилепсия. При проведении видео-ЭЭГ мониторинга в ходе сна зарегистрирована эпилептиформная активность в виде коротких (до 1 сек) разрядов генерализованной пик/полипик-волновой активности, с амплитудным преобладанием в лобных отделах.
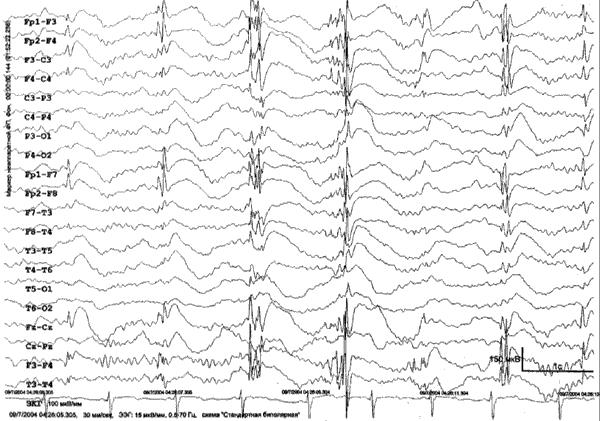
ЮМЭ имеет благоприятный прогноз: когнитивные нарушения у пациентов отсутствуют, и приступы в подавляющем большинстве случаев купируется на фоне терапии антиэпилептическими препаратами (АЭП). Вместе с тем, больные вынуждены принимать АЭП долгие годы, так как отмена терапии, даже при многолетней ремиссии, в высочайшем проценте случаев приводит к рецидиву приступов [1, 4, 11, 20]. Частота рецидивов после отмены лечения у больных ЮМЭ составляет, по данным разных авторов, от 50% до 100% [2].
Традиционно лечение ЮМЭ в ХХ веке осуществлялось препаратами вальпроевой кислоты [4, 9, 11, 27]. Однако, в последние годы были синтезированы новые высокоэффективные АЭП широкого спектра действия (ламотриджин, топирамат, леветирацетам), и в отдельных публикациях была показана их эффективность при ЮМЭ [6, 10, 14, 23, 24, 25, 29]. Кроме того, накопилось большое количество данных, свидетельствующих о недостаточной эффективности вальпроатов и их высокой токсичности, особенно у женщин [1, 9, 10, 12, 19]. Все это предопределило поиск оптимальных АЭП в лечении ЮМЭ.
Целью настоящего исследования было изучение эффективности и переносимости леветирацетама (кеппра, UCB) у больных юношеской миоклонической эпилепсией.
Нами обследовано 12 пациентов с установленным диагнозом ЮМЭ в возрасте 14–22 лет, 4 мужчин и 8 женщин. Кеппра назначалась в комбинации с другими АЭП (вальпроаты, суксилеп) в 3 случаях и при монотерапии — в 9 случаях (из них в 3 — в стартовой монотерапии). В 6 из 9 случаев монотерапии кеппра была назначена вместо препаратов вальпроевой кислоты (депакин). Анализировалось влияние кеппры на частоту и характер различных видов приступов, данные ЭЭГ, а также переносимость препарата. Всем больным до обследования и в динамике проводился видео-ЭЭГ мониторинг (ВЭМ). Катамнез составил от 7 мес. до 3 лет.
Результаты
Возраст дебюта приступов у обследованных больных варьировал от 7 до 16 лет (средний — 11,7 лет). Все 12 пациентов имели облигатный тип приступов при ЮМЭ — миоклониче-ские пароксизмы. Генерализованные тонико-клонические и клонико-тонико-клонические приступы констатировались в 9 случаях, и типичные абсансы — в 2 случаях. Также у 3 больных отмечался особый тип приступов — эпилептический миоклонус век.
Таким образом, исключительно миоклонические приступы были диагностированы лишь у двух пациентов, а сочетание МП, ГСП и абсансов — в одном случае. В большинстве случаев (8 больных) типичный фенотип ЮМЭ заключался в комбинации МП и ГСП. У 50% больных (6 случаев) отмечалась фотосенситивность как клиническая, так и по данным ЭЭГ. Во всех случаях приступы провоцировались депривацией сна.
У всех больных неврологическое обследование и ориентировочное нейропсихологическое тестирование не выявило отклонений от нормы. У 3 пациенток женского пола был диагностирован высокий уровень невротизации и склонность к депрессивным реакциям.
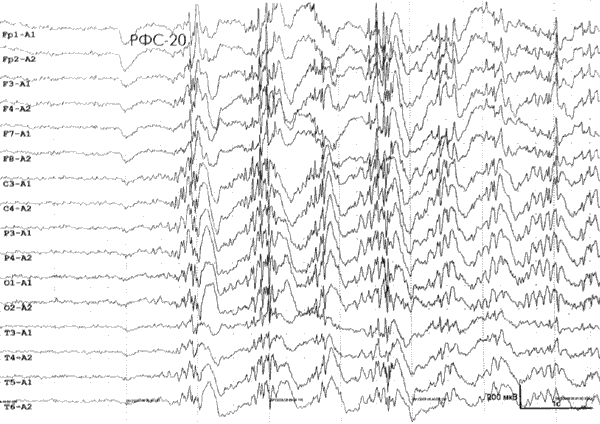
Во всех случаях, хотя бы однократно, при рутинном ЭЭГ-исследовании была обнаружена генерализованная (диффузная) эпилептиформная активность. Данная активность проявлялась, преимущественно, короткими разрядами генерализованных пик- или полипик-волновых комплексов в фоне, при ритмической фотостимуляции и/или в течение 3 сек после закрывания глаз. У 2 больных с абсансами на ЭЭГ констатировалась генерализованная высоко синхронизированная регулярная пикволновая активность частотой 3–4 Гц с продолжительностью разрядов максимально до 4 сек. У 6 больных эпилептиформная активность исключительно или преимущественно выявлялась при ритмической фотостимуляции на частотах 15–20 Гц и/или при закрывании глаз. Важно отметить, что именно в этой группе пациентов у 3 из 6 больных был выявлен эпилептический миоклонус век (рис. 2).
Рис. 2. Пациентка Г.А., 17 лет. Диагноз: Юношеская миоклоническая эпилепсия. При проведении видео-ЭЭГ мониторинга в ходе сна зарегистрирована эпилептиформная активность в виде коротких (до 1 сек) разрядов генерализованной пик/полипик-волновой активности, с амплитудным преобладанием в лобных отделах.
Терапевтический анамнез у пациентов был следующим. В 3 случаях кеппра назначалась как стартовая монотерапия при вновь диагностированной ЮМЭ. В 6 случаях кеппра применялась также в монотерапии при замене препаратов вальпроевой кислоты (депакин): в 3 случаях ввиду недостаточной эффективности (2 — продолжающиеся приступы и 1 — высокий индекс эпилептиформной активности в виде интериктальных разрядов на ЭЭГ) и в 3 —вследствие выраженных побочных эффектов по просьбе пациентов. В 3 оставшихся случаях кеппра была добавлена к другим АЭП (2 — депакин и 1 — суксилеп) ввиду недостаточной эффективности предшествующей терапии. Дозы кеппры у больных ЮМЭ варьировали от 1500 до 4500 мг/сут в 2 приема. Во всех случаях осуществлялась постепенная титрация дозы, занимавшая от 3 до 8 нед.
При обследовании больных в катамнезе анализировалось влияние кеппры на течение эпилепсии, данные ЭЭГ; а также переносимость препарата. Во всех 3 случаях стартовой монотерапии был получен положительный эффект, достигающий 100%: полное купирование всех типов приступов и постепенная нормализация биоэлектрической активности головного мозга по данным ВЭМ. У всех 6 больных, переведенных с монотерапии депакином на кеппру, эпилептических приступов не отмечалось. В 5 случаях констатировалось полное блокирование эпилептиформной активности по данным ВЭМ и в 1 — отсутствие эффекта в отношении эпилептиформных разрядов. У 3 больных кеппра была добавлена к суксилепу (1 случай) и депакину (2 случая). Эпилептические приступы были полностью купированы, в том числе, и резистентный ко многим другим АЭП эпилептический миоклонус век. Однако достоверное улучшение результатов ВЭМ по уменьшению интериктальной эпилептиформной активности было достигнуто лишь в 1 из 3 случаев. Таким образом, в общей группе больных стойкое купирование эпилептических приступов наблюдалось в 100% случаев, а полное блокирование или достоверное уменьшение индекса интериктальных эпилептиформных разрядов на ЭЭГ — в 75%. В 5 случаях из 6 кеппра достоверно уменьшила проявления фотосенситивности как по клиническим проявлениям, так и по данным ЭЭГ.
Побочные эффекты (ПЭ) терапии кеппрой были отмечены лишь у 1 пациентки (8%). Больная Г.А., 17 лет, с дебютом приступов в 9 лет, фенотипом МП + ГСП, принимала ранее фенобарбитал, клоназепам, суксилеп, депакин, топамакс в разных комбинациях. Эпилептические приступы удалось купировать при сочетании депакина 1750 мг/сут и топамакса 150 мг/сут. Однако эпилептиформная активность на ЭЭГ в виде частых генерализованных интериктальных разрядов полипик-волновых комплексов в фоне отмечалась постоянно, с высоким индексом. Кроме того, у больной были зарегистрированы различные ПЭ терапии: резкое снижение аппетита и массы тела, аменорея, гипохромная анемия, нейтропения. В связи с катастрофическим падением массы тела, наличием других ПЭ, а также неудовлетворительным результатом воздействия на эпилептиформную активность, было принято решение (совместно с семьей пациентки) изменить лечение. Был отменен топамакс, снижена доза депакина до 1000 мг/ сут и одновременно введена кеппра в конечной дозе 3500 мг/сут (титрование дозы проводилось в течение 3 нед.). Эпилептических приступов не возникло, и был констатирован положительный эффект в виде резкого уменьшения индекса интериктальной эпилептиформной активности на ЭЭГ. Однако, уже спустя 1 нед. после выхода на полную дозу кеппры у больной развился синдром насильственной нормализации Ландольта. Проявилось это резкой раздражительностью, злобностью, бессонницей, агрессией (вербальной и невербальной) в отношении родителей; выраженным снижением фона настроения. Доза кеппры была снижена до 2000 мг/сут. В настоящее время пациентка получает депакин хроно 1000 мг/сут и кеппру 2000 мг/сут. Приступов нет, побочные эффекты полностью купированы: восстановились вес и менструальный цикл, нормализовались показатели крови. Эмоционально лабильна, но старается избегать конфликтов, критична. Однако на ЭЭГ продолжает регистрироваться выраженная эпилептиформная активность в виде диффузных полипик-волновых разрядов в фоне интериктально.
Длительность катамнестического наблюдения за пациентами, принимавшими кеппру, составила от 7 мес. до 3 лет (в 92% случаев — более 1 года). Удержание на терапии в настоящий момент составляет 100%. Ни один из пациентов не прекратил прием кеппры в связи с неэффективностью, плохой переносимостью или по каким-либо другим причинам. Рецидив ГСП однократно наблюдался лишь у 1 больного при пропуске приема препаратов (суксилеп + кеппра) и после жесткой депривации сна. Также констатировался стойкий пролонгированный эффект кеппры в отношении блокирования интериктальной эпилептиформной активности на ЭЭГ.
Обсуждение
Современная история лечения ЮМЭ насчитывает 50 лет. D. Janz и W. Christian, описавшие это заболевание в 1957 году, впервые применили в терапии производные барбитуровой кислоты: фенобарбитал и примидон (гексамидин). Парадоксально, но полная ремиссия приступов была достигнута авторами в 86% случаев! Они также применили фенитоин, и установили, что данный препарат мало эффективен у больных ЮМЭ и вызывает аггравацию приступов в 33% случаев.
Таким образом, уже полвека назад было показано, что эпилептические приступы при ЮМЭ относительно легко купируются антиэпилептическими препаратами, в частности барбитуратами. Проблема заключалась в высокой частоте ПЭ барбитуратов, прежде всего, в отношении когнитивных функций и нейроэндокринной системы, особенно у мужчин [16].
С 80-х гг. прошлого века в клиническую практику прочно вошли препараты вальпроевой кислоты (конвулекс, депакин). Была показана высокая эффективность вальпроатов (VPA) в купировании всех видов приступов у больных ЮМЭ (миоклонус, ГСП, абсансы), и они прочно утвердились как препараты первого выбора в лечении данной формы эпилепсии [3, 9, 23, 27].Полная медикаментозная ремиссия при назначении вальпроатов достигается у 80–87% больных, причем в подавляющем большинстве случаев — на монотерапии [9, 15]. При недостаточной эффективности вальпроатов, они всегда оставались базовыми препаратами в комбинированной терапии: VPA + суксилеп (при резистентных абсансах); VPA + фенобарбитал (при резистентных ГСП); VPA + клоназепам (при выраженном миоклонусе и фотосенситивности) [27, 28].
Однако с накоплением большого клинического опыта применения производных вальпроевой кислоты за последние 20 лет стали очевидны серьезные проблемы. Во-первых, вальпроаты, высоко эффективные при миоклонусе и абсансах, имеют достоверно меньшую эффективность при ГСП и эпилептическом миоклонусе век [2, 5, 21]. Во-вторых, вальпроаты недостаточно эффективны в отношении блокирования интериктальной эпилептиформной активности на ЭЭГ. Именно при ЮМЭ важно добиваться полного блокирования эпилептиформных разрядов на ЭЭГ, так как их сохранение — один из важнейших факторов рецидива приступов при снижении дозы АЭП и отмене терапии [2]. И наконец, в-третьих (это наиболее важно!), накопились данные, свидетельствующие о высокой частоте серьезных побочных эффектов вальпроатов при длительной терапии [4, 20].
В нашей первой публикации, посвященной побочным эффектам АЭП в лечении идиопатической генерализованной эпилепсии [4], у 154 пациентов, принимавших препараты вальпроевой кислоты, различные ПЭ были констатированы в 49% случаев. В нашей публикации 2008 г. [10], у 100 больных эпилепсией, получавших монотерапию вальпроатами, побочные эффекты наблюдались в 62% случаев! Это касается, прежде всего, влияния вальпроатов на нейроэндокринную систему, функцию печени, косметических побочных эффектов [1]. Многие авторы считают крайне нежелательным назначение вальпроатов девушкам и женщинам детородного возраста [20]. Данные препараты могут приводить к ожирению, нарушению углеводного обмена (гипергликемия), расстройству менструального цикла, поликистозу яичников. Кроме того, показано, что по сравнению с другими АЭП, вальпроаты обладают более выраженным тератогенным эффектом [19].
Все вышесказанное предопределило поиск в XXI веке новых препаратов в лечении ЮМЭ — эффективных и относительно безопасных. В отдельных публикациях рассматривается возможность применения ламотриджина и топирамата при ЮМЭ [6, 13, 18, 29]. Ламотриджин, обладая хорошей переносимостью, недостаточно эффективен в монотерапии ЮМЭ; кроме того, в отдельных случаях может приводить к аггравации приступов, в частности, миоклонуса [13]. Топамакс высоко эффективен при генерализованных судорожных приступах, однако, при абсансах и миоклонусе его эффективность ниже. В целом, препарат показывает достаточно высокую результативность в лечении больных ЮМЭ и, безусловно, является перспективным [10].
С начала века в клинической практике начал применяться препарат широкого спектра действия — леветирацетам (кеппра). В настоящее время доказана высокая эффективность кеппры при большинстве эпилептических приступов у детей и взрослых, а также хорошая переносимость препарата [7, 20]. К 2008 г. накопились данные, свидетельствующие об избирательной эффективности леветирацетама при идиопатической генерализованной эпилепсии, в частности, при ЮМЭ. Sharpe и соавт. (2008) провели исследование леветирацетама в лечении юношеской миоклонической эпилепсии у 30 пациентов [24]. Препарат назначался в монотерапии (у 12 — стартовая терапия) в дозе 500–3000 мг/сут (10–59 мг/кг/сут). Средняя продолжительность лечения составляла 27 мес. В результате монотерапии леветирацетамом у 24 из 30 больных (80%) была достигнута стойкая медикаментозная ремиссия, и еще у 2 — существенное снижение частоты приступов. Авторы отмечают, что среди 20% больных, у которых ремиссии достигнуто не было, преобладали пациенты с атипичным течением заболевания. Кроме того, терапевтический эффект был хуже в небольшой группе больных с абсансными приступами. В результате исследования авторы получили еще один важный вывод: эффективность леветирацетама не зависит от эффекта предшествующей терапии. Большинство пациентов до назначения леветирацетама получали вальпроаты. ПЭ наблюдались лишь в 1 случае из 30 — «нарушение поведения».
В исследовании N. Specchio и соавт. (2008) изучалось влияние леветирацетама на интериктальную эпилептиформную активность и фотопароксизмальный ответ на ЭЭГ у больных ЮМЭ [25]. Обследовано 48 пациентов, из них 10 с впервые установленным диагнозом ЮМЭ. Средняя доза леветирацетама составляла 2200 мг/сут, а средний период наблюдения — 19,3 мес. До начала лечения межприступная эпилептиформная активность на ЭЭГ констатировалась у 91% больных, а фотопароксизмальный ответ — у 35%. На фоне терапии леветирацетамом полная нормализация ЭЭГ констатировалась в 56% случаев, а блокирование или выраженная редукция фотопароксизмальной реакции — в 76%. В результате исследования авторы сделали вывод о высокой эффективности леветирацетама в блокировании интериктальных эпилептиформных разрядов и фотопароксизмального ответа на ЭЭГ. Ранее Kasteleijn-Nolst Trenite D.G. и соавт. (1996) показали эффективность леветирацетама в блокировании фотосенситивности у больных эпилепсией [17].
Наше исследование включает лишь предварительные результаты, основанные на применении леветирацетама (кеппры) в лечении 12 больных ЮМЭ. В общей группе стойкое купирование эпилептических приступов было констатировано в 100% случаев, а полное блокирование или выраженное уменьшение индекса интериктальных эпилептиформных разрядов на ЭЭГ — в 75%. У 5 из 6 пациентов кеппра достоверно уменьшила проявления фотосенситивности, как по клиническим проявлениям, так и по данным ЭЭГ. Влияние кеппры на эпилептиформную активность было менее выражено в группе больных, получавших политерапию (комбинацию с вальпроатами или суксилепом) и при наличии абсансов (2 пациентов). Это согласуется с результатами исследования D.V. Sharpe и соавт. (2008), показавшего недостаточный эффект леветирацетама при атипичном течении ЮМЭ и наличии у больных абсансных приступов [24].
Нами показана высокая эффективность кеппры при особом типе приступов в рамках ЮМЭ — эпилептическом миоклонусе век. Данный тип приступов нередко возникает у больных с фотосенситивностью и провоцируется закрыванием глаз; нередко возникает аутоиндукция приступов [5]. Миоклонус век может сочетаться с другими типами приступов, в частности с абсансами и ГСП; отмечена его резистентность к большинству АЭП [2]. В публикации P. Striano и соавт. (2008) отмечается высокая эффективность леветирацетама в моно- и политерапии в лечении больных синдромом Дживонса — эпилептический миоклонус век с абсансами или без них. Положительный эффект леветирацетама констатировался у 80% из 35 больных синдромом Дживонса. Блокирование диффузной эпилептиформной активности на ЭЭГ, возникающей при закрывании глаз, отмечалось в 57% случаев [26].
Во всех публикациях показана хорошая переносимость леветирацетама, в том числе, и высоких доз препарата. У обследованных нами 12 больных ЮМЭ, принимавших кеппру, лишь в 1 случае отмечался ПЭ в виде феномена насильственной нормализации Ландольта. В исследовании Sharpe D.V. и соавт. (2008) ПЭ также наблюдались у 1 из 30 больных и проявлялись, по описанию авторов, «нарушением поведения» [24]. Таким образом, ПЭ леветирацетама констатируются очень редко [8]. Однако не следует забывать о возможности появления феномена насильственной нормализации у взрослых больных, принимающих леветирацетам, особенно в случаях высокой клинической эффективности препарата и при полном блокировании интериктальных эпилептиформных разрядов на ЭЭГ.
В заключении хотелось бы акцентировать внимание на то, что большинство пациентов с диагнозом ЮМЭ должны принимать АЭП очень длительно. Проблема заключается в высокой частоте рецидивов приступов после отмены АЭП. Отмена препаратов даже спустя 4–5 лет полной электроклинической ремиссии вызывает рецидив приступов не менее чем у 75% больных [5, 28]. При этом важно полное блокирование интериктальной эпилептиформной активности по данным ВЭМ, включая запись во сне [2]. По этой причине, многолетнее лечение должно проводиться высокоэффективным и хорошо переносимым АЭП, причем желательно — в монотерапии.
Проведенное предварительное исследование и данные литературы убедительно свидетельствуют, что леветирацетам (кеппра) может быть препаратом выбора в лечении юношеской миоклонической эпилепсии. Показано, что кеппра не уступает по эффективности вальпроатам и имеет существенные преимущества перед ними по критерию безопасности. Кеппра также высоко эффективна в блокировании интериктальной эпилептиформной активности на ЭЭГ и феномена фотосенситивности. Лучший эффект препарата достигается при монотерапии, особенно при стартовой. В настоящее время назначение вальпроатов в качестве базового препарата при многих формах эпилепсии вызвано не столько большей их эффективностью, сколько лучшей изученностью. Необходимы дальнейшие исследования по эффективности леветирацетама при идиопатической генерализованной эпилепсии и, в частности, при ЮМЭ.
Библиография
1. Воронкова К.В., Петрухин А.С., Пылаева О.А., Холин А.А. Рациональная антиэпилептическая фармакотерапия // Москва, Бином, 2008. — 192 с.
2. Миронов М.Б., Мухин К.Ю., Петрухин А.С., Холин А.А. Контроль эффективности лечения пациентов с юношескими формами идиопатической генерализованной эпилепсии и состояние «псевдоремиссии». — Журн неврол и психиат. — 2005. — Т. 105. — №8. — С. 24–28.
3. Мухин К.Ю., Никанорова М.Ю., Левин П.Г. Ювенильная миоклоническая эпилепсия // Журн неврол и психиат. — 1995. — Т.95. — N.3. — С. 17–21.
4. Мухин К.Ю., Петрухин А.С., Рыкова Е.А. Побочные эффекты антиконвульсантов при лечении идиопатиче-ской генерализованной эпилепсии // Журн неврол и психиат. — 1997. — Т. 97. — N.7. — С. 26–30.
5. Мухин К.Ю., Петрухин А.С. Идиопатические формы эпилепсии: систематика, диагностика, терапия. — М., 2000. — 319 С.
6. Мухин К.Ю., Глухова Л.Ю., Петрухин А.С., Миронов М.Б., Соборнова А.М. Топамакс при монотерапии эпилепсии // Журн неврол и психиат. — 2004. — Т. 104. — N. 8. — С. 35–40.
7. Мухин К.Ю., Пилия С.В., Чадаев В.А., Миронов М.Б., Пе-трухин А.С. Кеппра в лечении эпилепсии: эффективность и переносимость // Журн неврол и психиат. — 2005. — Т.105. — N. 1. — С. 49–51.
8. Мухин К.Ю., Тысячина М.Д., Миронов М.Б., Пилия С.В., Петрухин А.С. Кеппра в монотерапии эпилепсии: электро-клиническая эффективность и переносимость // Русский журнал детской неврологии. — 2007. — Т. 2. — № 3. — С. 14–23.
9. Мухин К.Ю., Петрухин А.С., Миронов М.Б., Долинина А.Ф. Вальпроат натрия (Депакин) в достижении ре миссии у больных идиопатической генерализованной 10.Мухин К.Ю., Тысячина М.Д., Мухина Л.Н., Петрухин А.С. Сравнительная эффективность и переносимость топирамата, вальпроатов и карбамазепина в монотера пии эпилепсии у детей и молодых взрослых // Рус. жур. дет. невр. — 2008. — Т. 3. — № 2. — С. 3–48.
11. Петрухин А.С., Мухин К.Ю., Медведев М.И. Основные принципы лечения эпилепсии у детей // Неврологический вестник — 1997. — Т. 29. — N 1–2. — C. 95 — 97.
12. Петрухин А.С., Демикова Н.С., Мухин К.Ю. Тератоген-ность антиэпилептических препаратов // Актуальные проблемы современной клинической генетики / ред. Г.Р. Мутовин, Л.Ф. Марченко. — Москва, 2001. — С. 205– 213.
13. Петрухин А.С., Мухин К.Ю., Калинина Л.В., Пылаева О.А. Ламиктал: поли- и монотерапия эпилепсии / Психиат и психофармакотер. — 2004 / приложение 1. — С. 20–26.
14. Gallagher M.J., Eisenman L.N., Brown K.M., Erbayat-Altay E., Hecimovic H., Fessler A.J., Attarian H.P., Gilliam F.G. Levetiracetam reduces spike-wave density and duration during continuous EEG monitoring in patients with idiopathic generalized epilepsy. // Epilepsia. — 2004. — V. 45. — P. 90–91.
15. Genton P., Gelisse P., Thomas P. Juvenile myoclonic epilepsy today: current definitions and limits // In: Eds. B. Schmitz, T. Sander / Juvenile myoclonic epilepsy: the Janz syndrome. — 2000. — Petersfield, WBP. — P. 11–32.
16. Janz D., Christian W. Impulsive petit mal // Dtsch. Z. Nervenheilk. — 1957. — V. 176. — P. 346–386.
17. Kasteleijn-Nolst Trenitй D.G., Marescaux C., Stodieck S., Edelbroek P.M., Oosting J. Photosensitive epilepsy: a model to study the effects of antiepileptic drugs. Evaluation of the piracetam analogue, levetiracetam. //Epilepsy Res. — 1996. — V. 25. — P. 225–30.
18. Kellett M.W., Smith D.F., Stockton P.A., Chadwick D.W. Topiramate in clinical practice: first year’s post-licensing experience in a specialist epilepsy clinic // J. Neurol. Neurosurg. Psychiatry. — 1999. — V. 66. — P. 759–763.
19. Meador K.J., Baker G.A., Finnell R.H. et al. In utero antiepileptic drug exposure. Fetal death and malformations // Neurology. — 2006. — V. 67. — P. 407–412.
20. Panayiotopoulos C.P. The epilepsies: Seizures, Syndromes and Management // Bladon Medical Publishing, 2005. — 540 P.
21. Panayiotopoulos C.P., Obeid T., Tahan R. Juvenile myoclonic epilepsy: a 5-year prospective study // Epilepsia. — 1994. — V. 35. — P. 285–296.
22. Panayiotopoulos C.P., Tahan R., Obeid T. Juvenile myoclonic epilepsy: factors of errors involved in the diagnosis and treatment // Epilepsia. — 1991. — V. 32 — P. 672–676.
23. Prasad A., Kuzniecky R.I., Knowlton R.C., Welty T.E., Martin R.C., Mendez M., Faught R.E. Evolving antiepileptic drug treatment in juvenile myoclonic epilepsy. // Arch Neurol. — 2003. — V. 60. — P. 1100–1105.
24. Sharpe D.V., Patel A.D., Abou-Khalil B., Fenichel G.M. Levetiracetam monotherapy in juvenile myoclonic epilepsy. // Seizure. — 2008. — V. 17. — P. 64–68.
25. Specchio N., Boero G., Michelucci R., Gambardella A., Giallonardo A.T., Fattouch J., Di Bonaventura C., de Palo A., Ladogana M., Lamberti P., Vigevano F., La Neve A., Specchio L.M. Effects of levetiracetam on EEG abnormalities in juvenile myoclonic epilepsy. // Epilepsia. — 2008. — V. 49. — P. 663–9.
26. Striano P., Sofia V., Capovilla G., Rubboli G., Di Bonaventura C., Coppola A., Vitale G., Fontanillas L., Giallonardo A.T., Biondi R., Romeo A., Viri M., Zara F., Striano S. A pilot trial of levetiracetam in eyelid myoclonia with absences (Jeavons syndrome). // Epilepsia. — 2008. — V. 49. — P. 425–30.
27. Sundqvist A., Nilsson B.Y., Tomson T. Valproate monotherapy in juvenile myoclonic epilepsy: dose-related effects on electroencephalographic and other neurophysiologic tests // Ther. Drug Monit. — 1999. — V. 21. — P. 91–96.
28. Thomas P., Genton P., Gelisse P., Wolf P. Juvenile myoclonic epilepsy // In: Epileptic syndromes in infancy, childhood and adolescence. 3th edition / Eds. J. Roger, M. Bureau, Ch. Dravet, P. Genton, C.A. Tassinari, P. Wolf — London: John Libbey, 2005. — P. 367–388.
29. Wallace S. Myoclonus and epilepsy in childhood: a review of treatment with valproate, ethosuximide, lamotrigine and zonisamide // Epilepsy Res. — 1998. — V. 29. — P. 147–154.
| Ноябрь 2013 г. |